Command Palette
Search for a command to run...
Published in Nature's Journal, a Wastewater Epidemiological Assessment Based on Gene Sequencing and Machine Learning Can Detect Viruses up to 4 Weeks earlier.

Over the past few years, global public health security has faced severe challenges. This is particularly true since the outbreak of the COVID-19 pandemic. Its pathogen, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has continued to evolve, with the emergence of multiple dominant variants. These variants possess varying abilities to infect and evade immune responses, significantly increasing the difficulty of epidemic prevention and control and the burden on healthcare systems.
Public health testing and SARS-CoV-2 genome sequencing are important means to comprehensively detect circulating variants.However, this type of clinical monitoring is often highly dependent on a large number of laboratory resources and requires individuals to actively participate in the test.It is difficult to fully track the emergence and spread of SARS-CoV-2 variants. Especially in areas with relatively limited medical resources or low willingness to test, clinical monitoring is more prone to detection bias, creating blind spots in prevention and control.
As a complementary approach, wastewater-based epidemiology (WBE) has played an important role in disease outbreak warning since it was first proposed in the 1940s to assess community infection. WBE mainly detects and tracks viral composition and dynamic changes by analyzing traces of viruses excreted by the human body in wastewater.Compared with clinical monitoring, WBE can objectively and unbiasedly reflect the group infection situation in the covered area without relying on individual active testing, achieve early warning, and has significant cost-effectiveness.
However, current mainstream wastewater monitoring methods (such as Freyja and COJAC based on linear regression) still have limitations.Detection needs to be based on the mutational pattern of known variants (such as reference sequences in the GISAID or UshER databases),If a new variant that has not been characterized or included in the clinical literature appears, it is often difficult to accurately identify it, which to a certain extent limits the detection efficiency of WBE.
To address this, a research team from the University of Nevada, Las Vegas proposed a multivariate analysis method called ICA-Var (Independent Component Analysis of Variants).The method is based on an unsupervised machine learning process design and uses Independent Component Analysis (ICA) to extract covariation and time-evolving mutation patterns from wastewater data.Earlier and more accurate variant detection is achieved.
Using this method, the research team accurately detected the Delta variant, the Omicron variant, and the recombinant XBB variant between late 2021 and 2023. This method not only reaffirms the effectiveness of wastewater monitoring for early warning of epidemic prevention and control, but also provides a new tool for comprehensively tracking viral mutations and spread in the absence of clinical monitoring.
The related research was published in Nature Communications under the title "Early detection of emerging SARS-CoV-2 variants from wastewater through genome sequencing and machine learning."
Research highlights:
* This method reveals the spatiotemporal dynamics of viral mutations in urban and rural areas, confirms the law of virus transmission from urban to rural areas, and provides an effective and low-cost variant detection paradigm for areas with poor medical access or lack of clinical sequencing data
* Compared with the current gold standard tool Freyja, ICA-Var's multivariate analysis method has significant advantages, and the detection time of Delta, Omicron and the latest EG.5, HV.1, BA.2.86 variants is an average of 1-4 weeks earlier

Paper address:
https://www.nature.com/articles/s41467-025-61280-5
Long-term, multi-point data collection
In this study, the wastewater samples used in the experiment were collected from August 2021 to November 2023.3,659 wastewater samples were collected from urban and rural areas in Southern Nevada.After collection, wastewater samples will be placed on ice on site and kept refrigerated until processing, for a storage time of no more than 36 hours.
During the nucleic acid extraction process,The research team first isolated nucleic acids from wastewater samples using the Promega Wizard Enviro Total Nucleic Kit (Cat. No. A2991) according to regulatory requirements. They then modified the Promega protocol, lysing the wastewater with a protease solution and using Macherey-Nagel NucleoMag Beads (Cat. No. 744970) to bind free nucleic acids. For RNA greater than 10 ng, the team used the New England BioLabs LunaScript RT SuperMix Kit for first-strand cDNA synthesis.
Sequencing library construction and sequencing,The research team used the CleanPlex SARS-CoV-2 FLEX Panel from Paragon Genomics to construct amplicon sequencing libraries, which were then sequenced on the Illumina NextSeq 500 or NextSeq 1000 platform using a 300-cycle flow cell.
In terms of sequencing data processing,The team first used cutadapt software (version 4.2) to remove Illumina adapter sequences from sequencing read pairs. They then mapped the sequencing read pairs to the SARS-CoV-2 reference genome (NC_045512.2) using bwa mem software (version 0.7.17-r1188). They then used the fgbio TrimPrimers tool (version 2.1.0, hard trimming mode) to remove Paragon Genomics CleanPlex SARS-CoV-2 FLEX amplicon primer sequences from the aligned reads. Finally, iVar variants software (version 1.4.1) was used to detect variants (based on allele frequency differences compared to the original 2020 reference genome), and samtools software (version 1.16.1) was used to calculate genome coverage and read depth.
After removing duplicate samples and positive/negative controls,The remaining 2,684 samples were used for quality control (QC) analysis.After rigorous quality control, only wastewater samples with a sequencing depth of 50x and covering at least 80% of the SARS-CoV-2 genome were retained for subsequent analysis, as shown in the figure below:
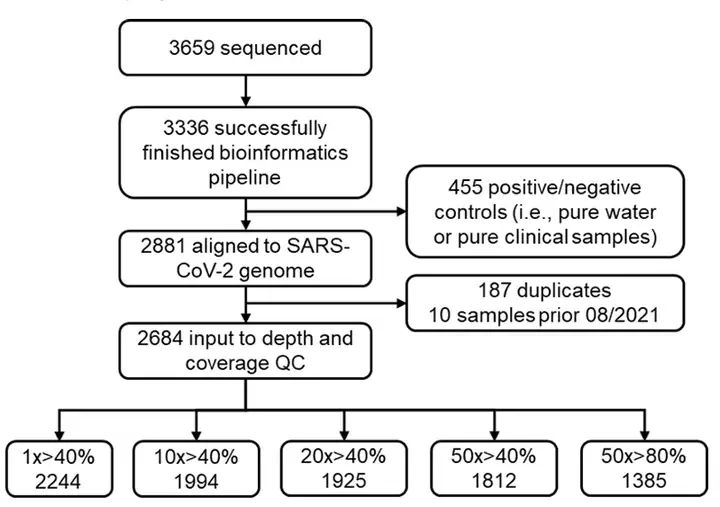
final,The study used 1,385 high-quality samples.Covering 59,422 mutation sites of SARS-CoV-2 variants for subsequent analysis.
To assist in verifying the effectiveness of the ICA-Var method, the research team used clinical data as a control and reference basis, and analyzed 8,810 high-coverage clinical SARS-CoV-2 sequence data from Nevada downloaded from the GISAID database, covering the period from September 2021 to November 2023.
With ICA as the core, a double regression method is introduced to create a new tool for COVID-19 detection
The core process of ICA-Var isIt processes the mutation frequencies in wastewater samples through independent component analysis and extracts independent co-variation mutation patterns.These patterns are then associated with the original samples through dual-regression to track virus variants, as shown in the figure below:
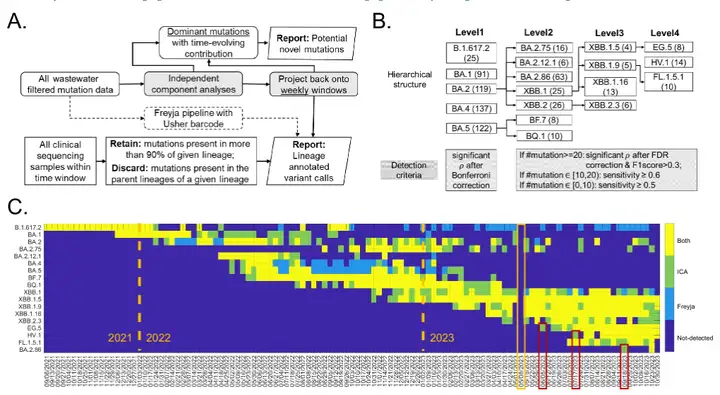
* A in the figure is the independent component analysis process.The two matrices are: weekly SARS-CoV-2 lineage detection (bottom row) and potential new mutations (top row)
* Figure B shows the hierarchical structure of 18 variants of concern.The main mutation sites of each variant (i.e., lineage-defining sites) were taken from http://covspectrum.org Summarized clinical data, with the number of major mutations in parentheses and the shaded boxes indicating the criteria to be tested in the proposed workflow.
*Figure C shows the comparison between the ICA-Var method and the state-of-the-art tool Freyja.For the newly emerged variants EG.5, HV.1, and BA.2.86, the red box indicates an earlier ICA-Var detection time; the yellow box indicates a week in which wastewater sampling was not performed due to technical problems.
Specifically, since the SARS-CoV-2 genome signal in wastewater samples is the result of a mixture of multiple variants and is interfered by sample degradation, sequencing errors, etc., traditional methods are difficult to directly analyze the characteristics of a single variant.The core idea of ICA-Var is to use independent component analysis——This blind source separation technology assumes that the mixed mutation signal is a linear combination of multiple "independent sources" and uses mathematical modeling to separate these independent patterns from the mixed data.
The research team first preprocessed the data.By performing quality control on SARS-CoV-2 genome sequencing data from wastewater samples, filtering out low-quality reads and noisy mutations, a "mutation frequency matrix" was constructed, with rows representing samples, columns representing mutation sites, and values representing the mutation frequency of each site in the sample. Independent component analysis was then performed on the mutation frequency matrix, decomposing the mixed signal into independent components. Each component represents a set of "covariation mutation patterns," or combinations of mutations characteristic of a particular variant that appear or disappear synchronously across samples over time.
here,The study used the Minimum Description Length (MDL) criterion to determine the number of independent components and performed independent component decomposition using the fastICA algorithm.To ensure the reliability of the results, they repeated the ICA analysis 50 times with different initial values, clustered and visualized the components obtained in each run with the help of ICASSO software, and finally retained only the reliable estimates corresponding to the tight clusters as the source matrix.
Afterwards, in order to further determine the weekly variant situation,The research team used the double regression method to re-project the source matrix obtained from independent component analysis into the original sample.Calculate the "contribution" of each independent component in each sample, that is, the relative abundance of the variant in the sample, so as to quantify the dynamic changes of different variants in time and space, such as the time of appearance, epidemic trends, and urban-rural distribution differences.
The research team used the full-sample source matrix as a set of source regressors in a general linear model (GLM) to find the signal decomposition patterns for each weekly sample related to the full-sample source matrix. They then used the signal decomposition patterns for each weekly sample as regressors in a second GLM to find the week-specific source matrix, still related to the full-sample source matrix. This process generated pairs of estimates that constituted the dual space and, together, provided the best approximation to the original full-sample independent component analysis source matrix in each weekly sample.
at last,The research team compared the isolated independent components with known variants in clinical sequencing data and annotated them.This can successfully determine the corresponding variant strain, or screen out unmatched covariation mutation patterns to warn of the possibility of new variant strains.
The ICA-Var method overcomes the drawbacks of traditional methods that rely on "predefined reference variant barcodes".By capturing the covariation patterns of mutations, it is possible to identify new variants earlier and more accurately than traditional methods.Combined with dual regression analysis, this method also reveals differences in urban and rural transmission and the temporal evolution of mutation sites. In summary, ICA-Var provides a more sensitive, comprehensive, and cost-effective tool for COVID-19 detection.
The detection efficiency exceeds the current gold standard tool Freyja and has the potential to predict new variants
To validate and evaluate the performance of ICA-Var, the research team compared it with the current gold standard tool Freyja, a tool for estimating the relative abundance of SARS-CoV-2 lineages in wastewater. It uses a "barcode" library consisting of mutations that define lineages to uniquely identify all known SARS-CoV-2 lineages, and uses a deep weighted, minimum absolute deviation regression method to solve for lineage abundance.Experiments have confirmed that the ICA-Var multivariate analysis method has more significant advantages.
As shown in the figure below, the model method and architecture section briefly explained that ICA-Var can detect new variants EG.5, HV.1, and BA.2.86 earlier, and the main content will be expanded in this section.
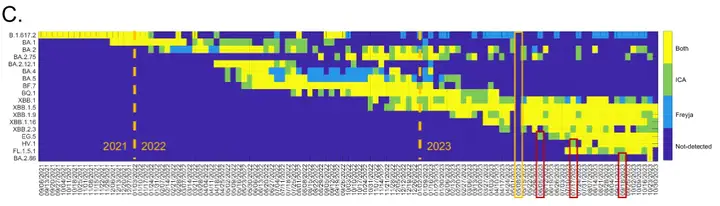
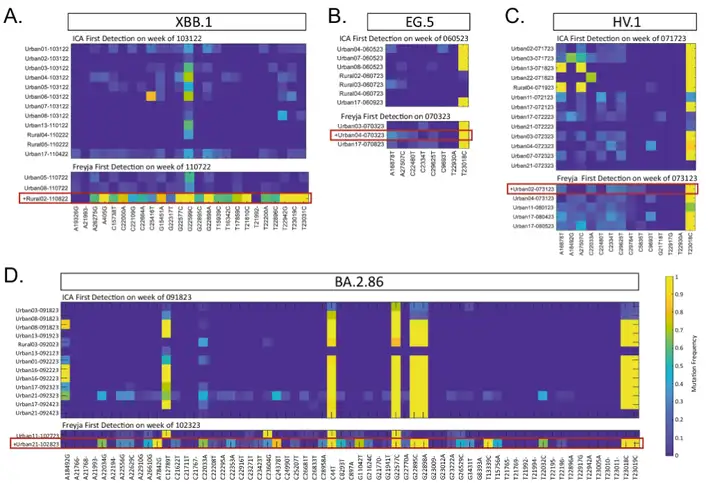
Specifically, in 2022,ICA-Var has been shown to detect variants such as BA.2, BA.4, BA.5, BF.7, BQ.1, XBB.1, and XBB.1.5 one or more weeks earlier than Freyja.In the detection of EG.5, ICA-Var detected this variant in the week of June 5, but Freyja did not identify the signal of EG.5 until July 3, when the abundance of wastewater samples reached 23.08%, and 5 of the 8 dominant mutation sites of EG.5 were already displayed. Similarly, for variants such as XBB.1, HV.1 and BA.2.86,ICA-Var was also detected several weeks earlier than Freyja.
This is due to ICA-Var integrating information from multiple samples on reliable but low prevalence mutation sites., improving statistical power and enabling earlier detection. This means it doesn't rely on a high proportion of dominant mutations in a single sample; it can enhance detection simply by aggregating weak signals from multiple samples. In contrast, Freyja requires at least one individual sample to clearly show a dominant mutation site to complete detection. This also means it relies more on a sufficiently strong mutation signal in a single sample and is less sensitive to weak or scattered signals.
The experiment further examined the dynamic trends of variants in urban and rural samples. Starting in early 2022, the research team sequenced and analyzed wastewater samples from rural areas in southern Nevada and conducted a comprehensive urban-rural epidemiological comparison, analyzing urban and rural samples separately on a weekly basis.
The results showed that among the 18 variants of concern, both ICA-Var and Freyja first detected 16 SARS-CoV-2 variants in urban wastewater samples before being found in rural samples, indicating that virus variants usually appear in cities first and then spread to rural areas. As shown in the figure below:
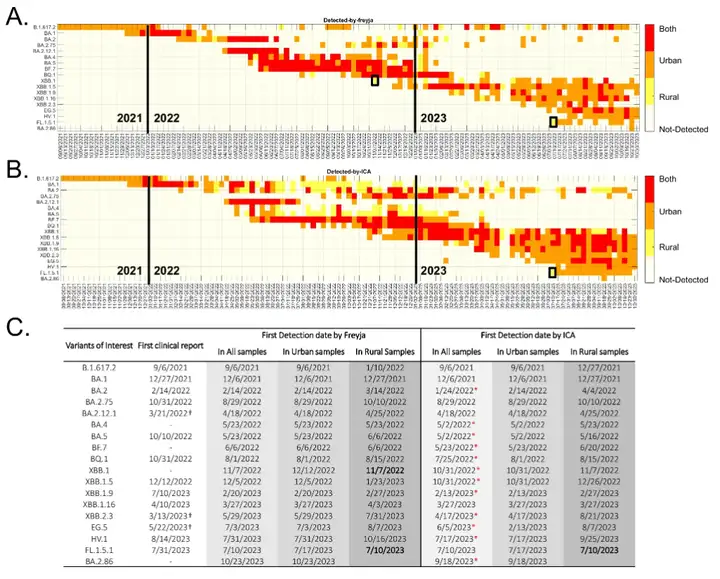
The exception is that Freyja initially detected XBB.1 in rural wastewater samples, while ICA-Var discovered the variant in urban wastewater samples a week earlier; both tools found FL.1.5.1 in rural wastewater samples, while the frequency and prevalence of the alternative allele of the dominant mutation of this variant were much lower in urban wastewater samples during the same period.
The study also revealed the temporal evolutionary trends of mutation sites. The research team compared 177 mutation sites with significant temporal evolutionary contributions between August 2021 and November 2023 with the dominant mutation sites of the B.1.617.2, BA.1, and XBB.1 variants, as shown in the figure below:
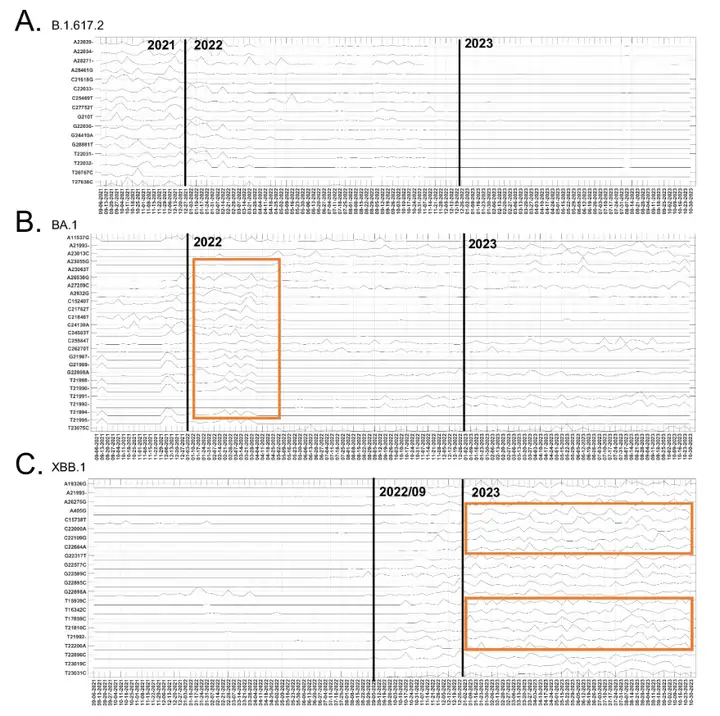
Of the 25 major mutation sites in the Delta variant (B.1.617.2), 16 showed significant fluctuations in contribution at the end of 2021, followed by a gradual decline in 2022. The contribution of related mutations in the Omicron subtype BA.1 increased significantly at the end of 2021 and peaked in early 2022. The contribution of some BA.1 mutation sites continued to fluctuate in 2023 and was found in other Omicron sublineages, such as XBB.1. Of the 25 major mutations in the XBB.1 variant, 22 showed significant temporal dynamic contributions, with a significant impact after September 2022. Multiple mutation sites exhibited similar fluctuation patterns, indicating co-variation, reflecting the recombination characteristics of XBB.1.
These analyses demonstrated that the temporal evolutionary contributions of mutation sites identified by ICA-Var were consistent with the clinical findings of Delta, Omicron, and XBB.1 variants, further illustrating the reliability of ICA-Var results and demonstrating its potential to identify novel mutational patterns that may lead to the emergence of new variants.
The experiment conducted a detailed verification of this. The research team screened 113 potential new mutation sites by cross-comparing them with the dominant mutation sites of 15 major variants. They then used a hierarchical clustering algorithm to classify these mutation sites into six characteristic clusters. As shown in the figure below:

Among these characteristic clusters, the mutation sites of 4 of them (clusters 2-5) overlap with the variants that appeared at the end of 2023. Cluster 1 and Cluster 6 have no overlapping mutations with known mutation sites. Among them, the mutation sites of Cluster 1 showed an obvious co-variation pattern after August 2023. The clinical sequencing data of GISAID showed that 8 of the mutation sites were verified and found to have a low reporting frequency in clinical samples. Therefore,These mutations may lead to the emergence of new coronavirus variants, which need to be further verified by clinical tests.Close monitoring is required.
Powered by machine learning, wastewater monitoring continues to evolve to drive high-quality virus prevention and control
As mentioned at the beginning, WBE is not a new method. As early as the 1940s, environmental virologists recognized the value of obtaining poliovirus through cell culture experiments in wastewater. Since then, WBE has been continuously improved and has become an effective tool for early warning of disease outbreaks.Since the outbreak of the COVID-19 pandemic, WBE has once again played a positive role in epidemic prevention and control.
For example, at the end of 2023, there were reports that a Swedish research team successfully detected the emergence of the new SARS-CoV-2 BA.2.86 variant by integrating genomic testing of sewage and COVID-19 cases. In addition, in order to more effectively and actively use WBE for the detection of new coronavirus variants, many laboratories have also developed or improved related models to provide more cost-effective tools for WBE.
For example, researchers from Tsinghua University, Hebei University of Science and Technology, and the Tianjin Ecological and Environmental Monitoring Center jointly published a study titled "Validation of methods for enriching and detecting SRAS-CoV-2 RNA in wastewater." The study compared two enrichment techniques, ultrafiltration and covalent affinity resin separation, with two detection methods, reverse transcription quantitative PCR (RT-qPCR) and reverse transcription digital PCR (RT-dPCR), to evaluate their performance in wastewater virus monitoring.
at last,The study indicated that the reverse transcription digital PCR (RT-dPCR) method is a better choice for detecting low concentrations of SARS-CoV-2 RNA in wastewater.The detection rate is higher and it has better tolerance to PCR inhibitors.
* Paper address:
https://link.springer.com/article/10.1007/s10311-025-01843-6
In addition, the team led by Professor Xingfang Li of the Department of Pathology and Laboratory Medicine at the University of Alberta, Canada, published a study titled "Quantification and Differentiation of SARS-CoV-2 Variants in Wastewater for Surveillance." Based on the Gamma (ABG) and Delta multiplex RT-qPCR detection methods previously developed for clinical samples, they targeted the Omicron subvariant and utilized its unique mutations.An Omicron triplex RT-qPCR assay was developed that can distinguish five major sublineages of Omicron variants.This is the first study to use a single-tube RT-qPCR triplex assay to detect and identify all Omicron subvariants in wastewater samples over a one-year period.
* Paper address:
https://pubs.acs.org/doi/10.1021/envhealth.3c00089
In short, the world today faces severe public health and safety challenges, and wastewater monitoring, as a highly effective means of population monitoring, is playing an irreplaceable role. With the continuous advancement of technology, wastewater monitoring will continue to evolve, from early targeted detection relying on known mutation patterns to breakthroughs in whole genome sequencing and identification of unknown pathogens. With continuously increasing sensitivity and coverage, wastewater monitoring will provide more accurate and critical data for epidemic warning, tracing, and policy making, becoming a vital supplement to the public health and safety defense line.
References:
1.https://www.nature.com/articles/s41467-025-61280-5
2.https://mp.weixin.qq.com/s/ZzzZt-uNNc5DsD-ib3Ww8g








