Command Palette
Search for a command to run...
Supporting Protein generation/folding/reverse Folding, Hunan University/University of Chinese Academy of Sciences/ByteDance Proposed the APM Model to Achieve all-atom Design and Function Optimization
As the main executor of life activities, proteins often realize their functions in the form of multi-chain complexes. From antibody-antigen recognition to enzyme-substrate binding, the precise interaction between multi-chain proteins is the core of understanding the mechanism of life.However, the current AI-driven protein modeling field shows a significant "single-chain bias". Although models such as AlphaFold and the ESM series have made breakthrough progress in the folding and design of single-chain proteins, the modeling of multi-chain complexes is still in its infancy.
Existing methods for processing multi-chain proteins generally adopt the "pseudo sequence connection" strategy, forcing multi-chains to be treated as single chains.This method severely limits the natural expression of interchain interactions - in real biological complexes, the atomic-level interactions between the spatial positions of interchains and the binding interface (such as hydrogen bonds and hydrophobic interactions) cannot be accurately modeled through linear connections. In addition, the generation of all-atom structures faces dual challenges: the complex conformation of amino acid side chains and the strong sequence-structure dependence make the de novo design of multi-chain complexes a difficult problem in the field.
To fill this research gap, Hunan University, University of the Chinese Academy of Sciences, and ByteDance Seed team proposed APM (All-Atom Protein Generative Model), an all-atom protein generation model designed specifically for multi-chain protein complexes. APM can not only directly generate multi-chain complexes with all-atom structures, but also supports basic tasks such as folding and reverse folding, and demonstrates excellent performance in the design of functional proteins such as antibodies and peptides.
The research results were selected for ICML 2025 under the title "An All-Atom Generative Model for Designing Protein Complexes".
Research highlights:
* Multi-chain native modeling: abandon pseudo-sequence connections and directly learn the atomic-level interactions between the independent spatial distribution of multi-chains and the binding interface;
* All-atom representation optimization: balance computational efficiency and structural details, and achieve atomic-level structure generation through the joint representation of amino acid type, backbone framework and side chain torsion angle;
* Sequence-structure dependency reinforcement: Maintaining the deep association between sequence and structure by decoupling the noise process and bidirectional task training (folding/unfolding).

Paper address:
Follow the official account and reply "APM" to get the complete PDFAPM protein generation dataset:
More AI frontier papers:
Dataset: Rich samples from single chain to multiple chains
APM is trained based on a carefully constructed multi-source protein dataset that integrates the structure and sequence information of single-chain and multi-chain proteins, providing rich learning materials for the model.
The single-chain dataset provides a rich foundation for in-chain modeling through multi-source fusion and quality filtering. It contains 187,494 samples in total, covering a wide range of protein types and functional categories. Its data mainly comes from 3 authoritative databases:
* PDB database: After the MultiFlow data processing process, 18,684 samples were screened;
* Swiss-Prot database: selected high-quality structures with pLDDT>85, and obtained 140,769 samples;
* AFDB database: Using stricter screening criteria, samples with pLDDT>95 were retained, totaling 28,041 samples.
The multi-chain protein dataset contains a total of 11,620 samples, covering protein complexes of 2-6 chains, providing key data support for multi-chain modeling. The multi-chain protein data comes from PDB biological assembly data (Biological Assemblies). To avoid information leakage in downstream tasks, the research team excluded three types of samples: samples that exist in the SAbDab antibody database; samples containing chains with a length of less than 30 (considered as peptides); samples with a length of more than 2,048 or lacking cluster IDs.
To improve the generalization ability of the model, the researchers randomly trimmed the multi-chain samples during the training process: for samples with more than 384 residues, the nearest 384 amino acids were retained, centered on the residue pairs at the inter-chain binding interface.This pruning strategy ensures that the model can focus on the key binding areas while avoiding memory overflow issues.In addition, the researchers mixed single-chain and multi-chain data in proportion, using the richness of single-chain data to improve the intra-chain modeling capabilities. Each sampling location is attached with rich metadata, including geographic location (interchain interaction site), structural properties (such as secondary structure type), and sequence features (amino acid type and conservation). This information provides multi-dimensional clues for the model to learn the mapping relationship between sequence, structure, and function.
APM protein generation dataset:
Model architecture: a three-module collaborative all-atom generation framework
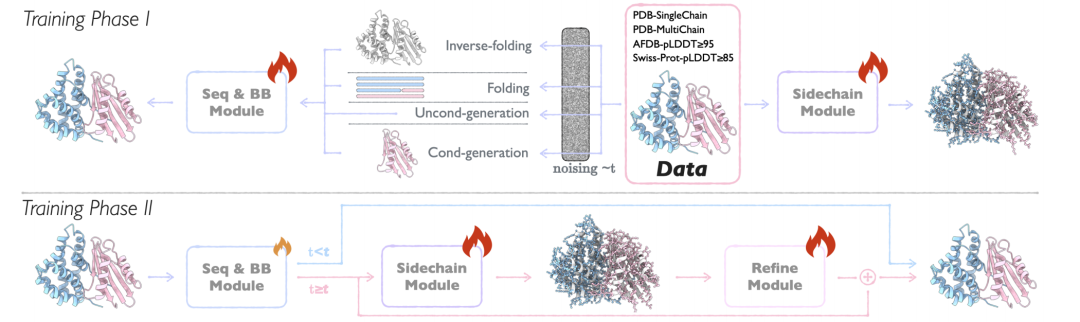
The core architecture of APM is composed of three modules with clear functions: Sequence and Backbone Generation Module (Seq&BB Module), Sidechain Generation Module (Sidechain Module) and Refine Module.Through innovative design, end-to-end generation from sequence to all-atom structure is achieved, while supporting various design tasks of multi-chain proteins.
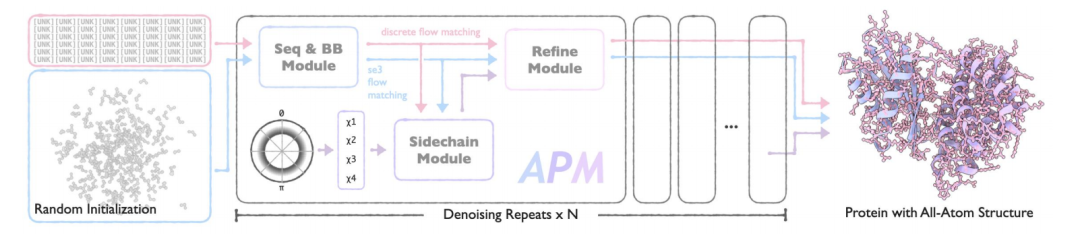
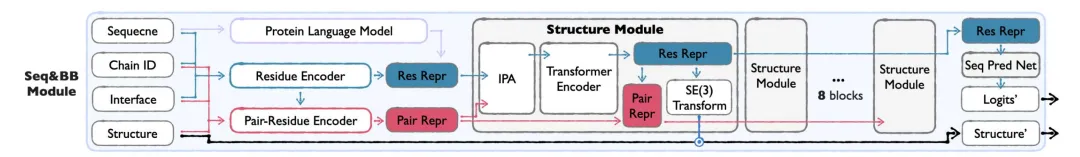
Seq&BB Module
This module is the basis of APM. It adopts the flow matching method to realize the joint generation of sequence and protein backbone, and can handle sequence-structure collaborative modeling tasks at the residue level.By decoupling the noise process of sequence and structure, the damage to the sequence-structure dependency relationship is reduced, and the folding/reverse folding tasks are performed with a probability of 50% to strengthen the bidirectional dependency learning. The core innovation of the module is:
* Decoupling noise process:Separating the sequence and structure noise processes avoids the destruction of inter-modal dependencies in traditional methods. The noise sequence and the noise backbone are sampled independently at different time steps, ensuring that the model can learn bidirectional sequence-structure dependencies.
* SE(3) Flow Matching:In view of the spatial transformation characteristics of protein backbone, three-dimensional special Euclidean group (SE(3)) flow matching is introduced to handle the translation and rotation parts separately.
* Multi-task learning:It also supports unconditional generation, conditional generation, folding and reverse folding tasks, and improves the generalization ability of the model through mixed task training. The loss function includes flow matching loss and consistency loss to ensure the smoothness of the generated trajectory.
Sidechain Module
To achieve all-atom structure generation, Sidechain Module predicts the conformation of amino acid side chains based on the sequence and backbone generated by Seq&BB.
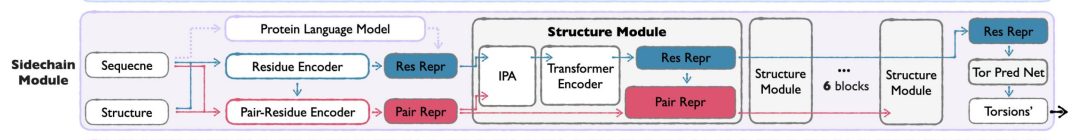
The module adopts the following strategies:
* torsion angle means:The side chain structure is parameterized by side chain torsion angles (up to 4 rotatable bonds), balancing computational efficiency and atomic-level detail, avoiding the complexity of direct modeling of all-atom coordinates.
* Two-stage training:The first stage focuses on the side chain packing task and learns the distribution of real side chain conformations; the second stage switches to reconstructing real side chains from predicted structures to ensure the applicability of the model in the generation scenario.
* Lightweight Design:Compared with Seq&BB Module, Sidechain Module uses fewer structural blocks and smaller hidden dimensions.
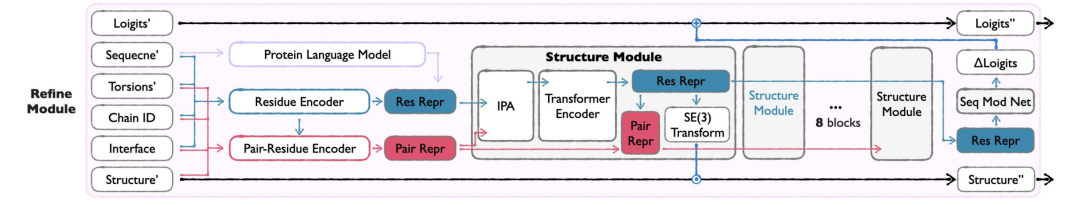
Refine Module
As the last link of APM, Refine Module integrates the output of Seq&BB and Sidechain Module, optimizes the sequence and backbone by correcting the loss, reduces atomic conflicts and improves structural rationality.The full-atom information is used to optimize the sequence and main chain structure, resolve structural conflicts, and make the generated results closer to the natural protein. This module is only activated in the late generation period (t≥0.8) to ensure that the input quality is sufficient to support the optimization.
Experimental conclusion: Multi-dimensional verification of APM's breakthrough performance
APM's experimental verification covers single-chain basic tasks, multi-chain core tasks and downstream functional design, and the results are all excellent.
Single-chain protein task: basic capabilities comparable to professional models
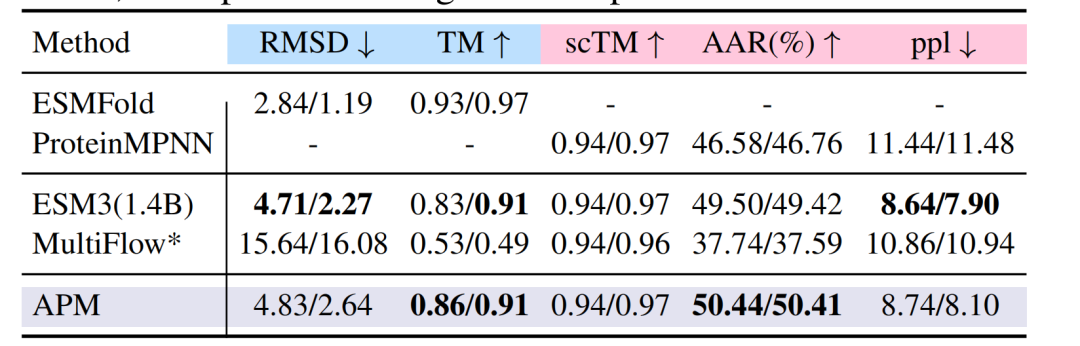
In the folding task, on the PDB dataset,The RMSD of APM is 4.83/2.64,The TM-score reached 0.86/0.91, which is comparable to the performance of ESM3, MultiFlow and other models; in the reverse folding task, the amino acid recovery rate (AAR) reached 50.44%, surpassing ProteinMPNN's 46.58%.
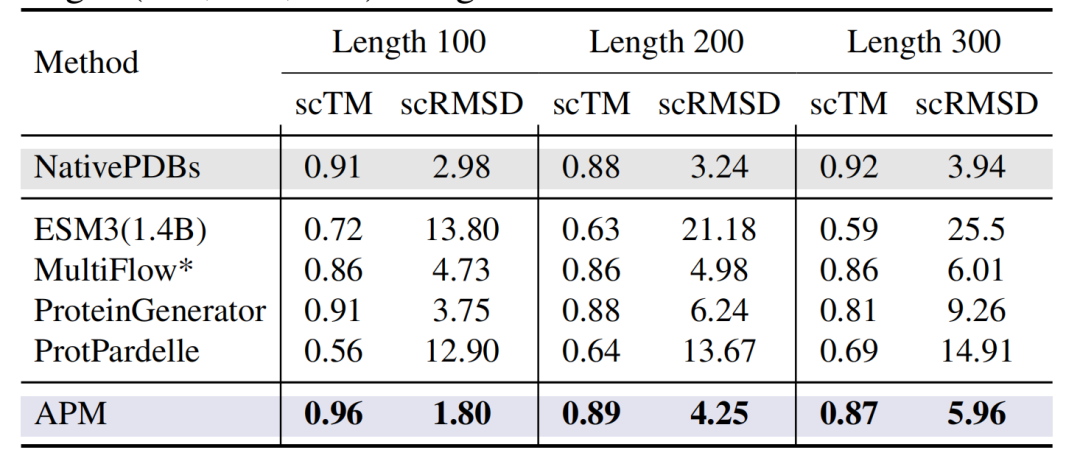
In addition, as shown in the figure below, in the unconditionally generated proteins with lengths of 100-300 residues,The scTM of APM is as high as 0.96 (Length 100), and the scRMSD is as low as 1.80.Significantly better than all-atom design models such as ESM3 (1.4B) and ProtPardelle.
Multi-chain protein tasks: the core advantage of native modeling
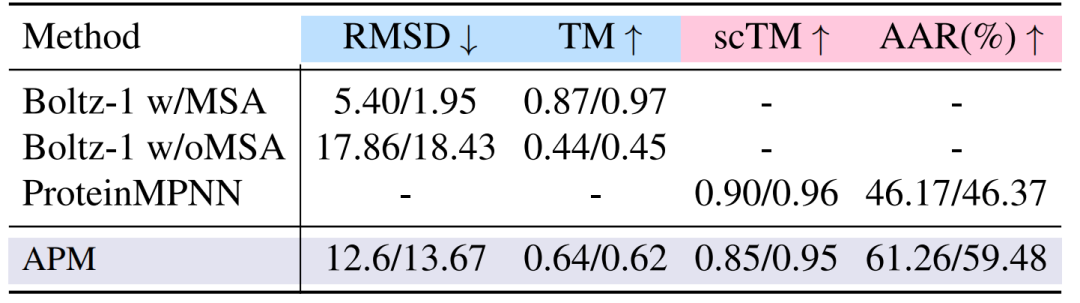
In the folding and unfolding experiments,On the 2-6 chain complex, the folding performance of APM is 12.6/13.67, which is lower than Boltz-1, but significantly exceeds Boltz-1 without MSA; the reverse folding scTM reaches 0.85/0.95, close to Boltz-1 with MSA, proving the validity of sequence-structure association. The experimental results are shown in the figure below.
Secondly,The multi-chain complex has a strong binding affinity.Taking the chain length of 50-100 as an example, the binding energy ΔG_RAA after all-atom relaxation reaches -112.65/-116.98, which is significantly better than Chroma (-83.96/-86.66) and APM_BB (-114.94/-114.45) using only the main chain, proving the necessity of all-atom information for modeling inter-chain interactions.
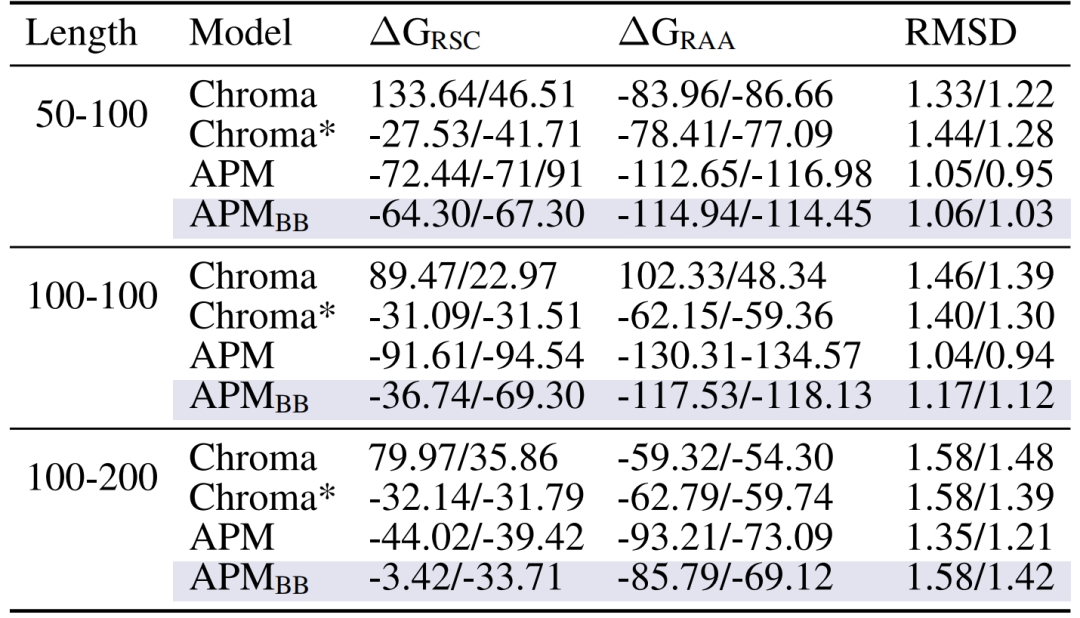
Interchain binding affinity between the generated complexes
Downstream functional design: breakthroughs in the application of antibodies and peptides
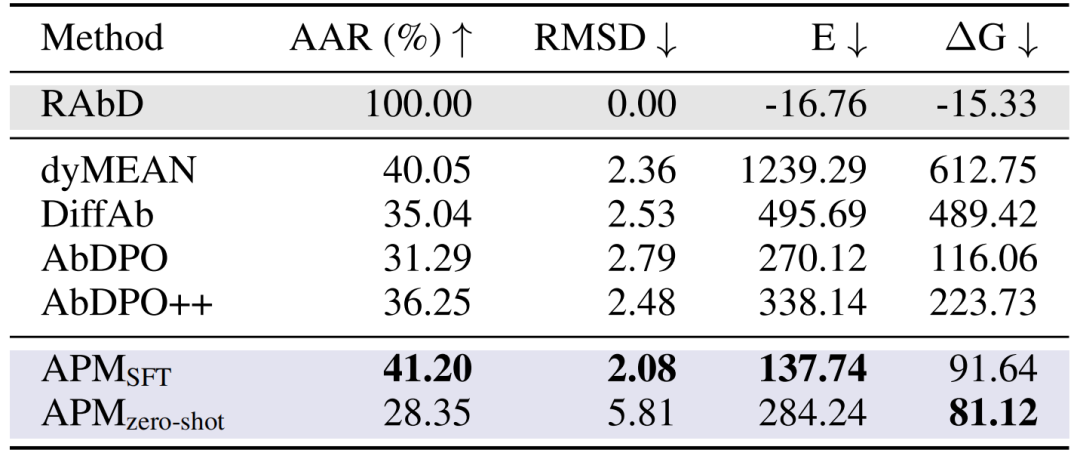
Antibody CDR-H3 Design:In the RAbD benchmark test, APM's AAR reached 41.20%, RMSD was 2.08, and binding energy ΔG was 91.64, surpassing methods such as dyMEAN and DiffAb. Although the sequence of the antibody generated by zero sample is very different from the natural one, the binding energy is better (ΔG 81.12), proving its universal binding ability.
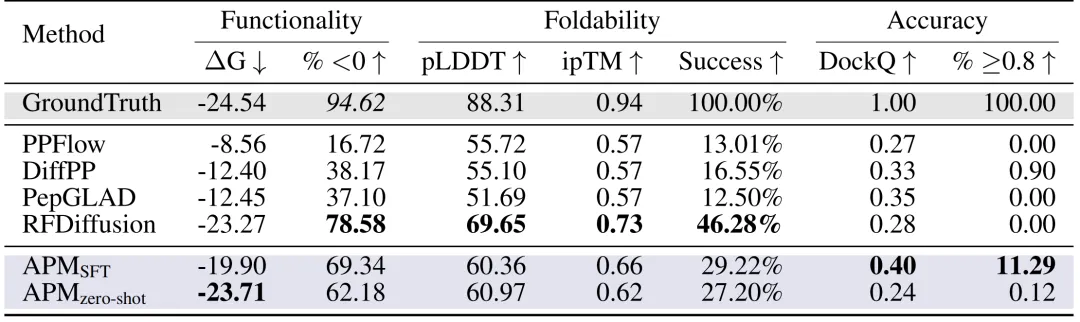
Peptide design:On the PepBench and LNR datasets, the researchers comprehensively evaluated the peptide design methods from three key aspects: functionality, foldability, and accuracy. As shown in the figure below, the binding energy ΔG of APM (SFT) reached -19.90, 69.34% samples had ΔG < 0, and the proportion of DockQ ≥ 0.8 reached 11.29%, far exceeding PPFlow, PepGLAD and other methods, and the folding stability (pLDDT 60.36, ipTM 0.66) was excellent.
Industry-R&D collaboration drives breakthroughs in all-atom protein generation technology
In the frontier biological field of all-atom protein generation, academia and business communities have never stopped exploring it, and a series of breakthrough results continue to attract attention.
In the academic world, AlphaFold3 launched by the DeepMind team has demonstrated strong capabilities in the field of all-atom protein generation by integrating multi-scale structural information with evolutionary sequence data.Achieved accurate modeling of complex protein folding patterns,Especially in the task of generating all-atom complexes containing cofactors and metal ions, the structural accuracy and energy rationality have been significantly improved compared with traditional methods. ESM-IF1 developed by the Stanford University research team takes a different approach. Based on an implicit folding model trained with massive evolutionary sequence data, it can directly generate all-atom protein structures with natural conformational characteristics, and performs outstandingly in the precise construction of enzyme active centers.
The business community is also actively deploying in this field, promoting industrial applications with technological innovation. Beijing Bio-Geometry Biotechnology Co., Ltd. released the world's first full-scenario atomic-level protein model - GeoFlow V2, which built an end-to-end diffusion generation framework that can achieve precise regulation of protein atoms. In the full-atom design of antibody CDR regions,It can optimize affinity and stability at the same time, significantly improving drug development efficiency.Insilico Medicine, an American biotechnology company, has developed a protein generation system that focuses on drug target protein design. The multi-constraint generation strategy it adopts can directionally optimize the binding sites between proteins and small molecule drugs while ensuring the rationality of the all-atom structure, providing a solid foundation for the efficient screening of candidate drugs.
These theoretical breakthroughs in academia and application innovations in the business community,Together, we will push all-atom protein generation technology from the laboratory to industrial practice, providing core support for breakthroughs in precision drug development, new biocatalyst design, and synthetic biology, and will hopefully create huge value in disease treatment and biomanufacturing in the future.
Reference Links:
1.https://mp.weixin.qq.com/s/a0bl9ek90t_-y8wy69Yu6Q
2.https://mp.weixin.qq.com/s/P-5o-R1qZY52Pq1yK5j6cQ