Command Palette
Search for a command to run...
Covering 200 Million Molecular Mass Spectra, the Czech Academy of Sciences Released the DreaMS Model to Build the world's Largest Mass Spectrometry Dataset GeMS
According to statistics, the natural small molecule chemical space currently explored by humans is less than 10% of its total amount, while in non-targeted metabolomics experiments, more than 90% of mass spectra have become "data wastes" due to the lack of reliable annotations.
In this crucial battle to decipher molecules, the core challenge lies in cracking the complex language of tandem mass spectrometry (MS/MS). As a cutting-edge tool for modern chemical analysis, the liquid chromatography-tandem mass spectrometry (LC-MS/MS) system achieves efficient separation of molecules through liquid chromatography, and then uses collision-induced dissociation technology to generate mass spectra of fragment ions. This process is similar to disassembling a molecule and analyzing its fragment puzzle.
However, existing analytical tools have significant limitations in piecing together a complete molecular picture:Even the advanced SIRIUS algorithm is overly dependent on a limited spectral library and artificial rules.When faced with unknown natural molecules that account for more than 80% in total, there is often a dilemma of having no library to check. A study published in Nature Methods in 2023 pointed out that in the global metabolomics database, only 2% MS/MS spectra were successfully annotated, and the remaining 98% were like reefs in the deep sea, seriously hindering the progress of new drug discovery and disease diagnosis research.
In order to solve this problem, a research team from the Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences drew on the breakthroughs made by the GPT series in the field of language and is committed to creating a dedicated translator for mass spectra. The researchers mined 700 million MS/MS spectra from the Global Natural Products Social Molecular Network (GNPS), successfully built the largest mass spectrometry dataset in history, GeMS, and trained a Transformer model DreaMS with 116 million parameters. This model is like letting artificial intelligence learn the "broken grammar" of molecules from scratch. By predicting masked spectral peaks and chromatographic retention order, it successfully discovered hidden structural patterns in unlabeled mass spectra:The 1,024-dimensional characterization vector it generates can accurately reflect the structural similarities between molecules and show strong robustness to signal fluctuations under different mass spectrometry conditions.
Research shows thatThe fine-tuned DreaMS performs well in a variety of mass spectrometry annotation tasks.Including predicting spectral similarity, molecular fingerprints, chemical properties, and the presence of fluorine, all of which surpass traditional algorithms and recently developed machine learning models., DreaMS has integrated 201 million spectra to build a super molecular network covering bacteria, plants, and human metabolites,It has created a "molecular encyclopedia" for the chemical community that can be updated in real time, providing extremely valuable resources for research and applications in related fields.
The relevant research results have been published in the internationally renowned journal Nature Biotechnology under the title "Self-supervised learning of molecular representations from millions of tandem mass spectra using DreaMS".

Paper address:
More AI frontier papers:
Download address of GeMS chemical mass spectrometry dataset:
https://go.hyper.ai/IC2yw
GeMS Dataset: 700 Million Spectra to Build a Mass Spectral Database
The core data foundation of this study is the GeMS data set deeply mined from the MassIVE GNPS repository, whose scale and quality are groundbreaking in the field of metabolomics.
Download address of GeMS chemical mass spectrometry dataset:
https://go.hyper.ai/IC2yw
As shown in the figure below,The research team integrated 250,000 LC-MS/MS experimental data covering biological and environmental fields, extracted approximately 700 million MS/MS spectra from them, and divided them into three subsets: GeMS-A, GeMS-B, and GeMS-C through strict quality control algorithms.Among them, GeMS-A mainly collects spectra with the 97% Orbitrap mass spectrometer, representing the highest quality standard; GeMS-C incorporates 52% Orbitrap and 41% QTOF spectra, greatly expanding the data scale while ensuring a certain quality. This hierarchical design not only retains the reliability of high-precision instrument data, but also covers a wider range of mass spectrometry technology sources through more inclusive subsets, ensuring the diversity of data sets.
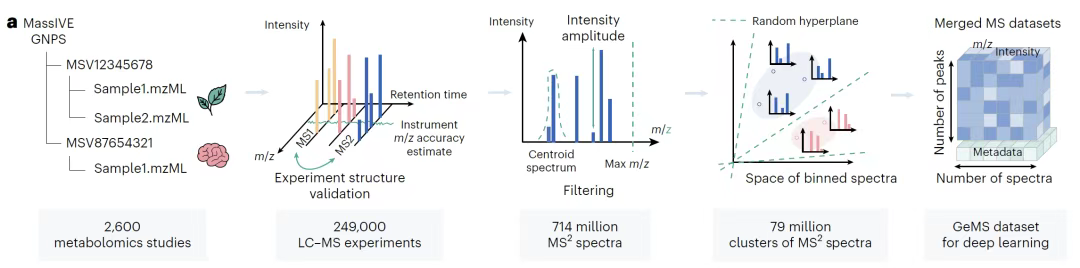
To solve the redundancy problem in large-scale data, the research team used the locality sensitive hashing (LSH) algorithm to efficiently cluster similar spectra, and generated nine variants by limiting the number of spectra in the cluster, optimizing computational efficiency while maintaining data representativeness. The GeMS dataset was finally stored in the compact HDF5 binary format.Convert the raw spectrum into a numerical tensor of fixed dimension,It breaks the scale bottleneck of traditional spectral libraries - as shown in the figure below, its data volume is several orders of magnitude larger than existing libraries, and its structure is highly standardized, providing unprecedented training materials for deep learning models. These data characteristics make GeMS the first ultra-large-scale mass spectrometry dataset suitable for unsupervised/self-supervised learning. It not only lays the foundation for the pre-training of the DreaMS model, but also provides data support with both accuracy and breadth for subsequent spectral similarity analysis, molecular structure characterization and other tasks through quality stratification and format optimization, promoting the metabolomics research from the traditional model that relies on limited reference libraries to the intelligent analysis paradigm based on massive raw spectra.
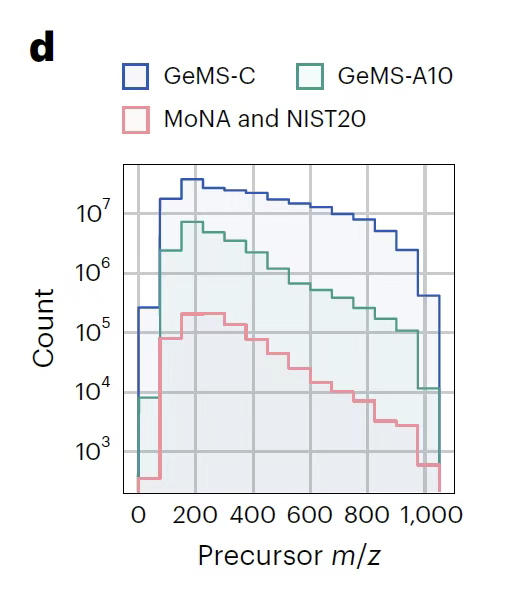
DreaMS model: a new paradigm for mass spectrometry analysis based on self-supervised Transformer
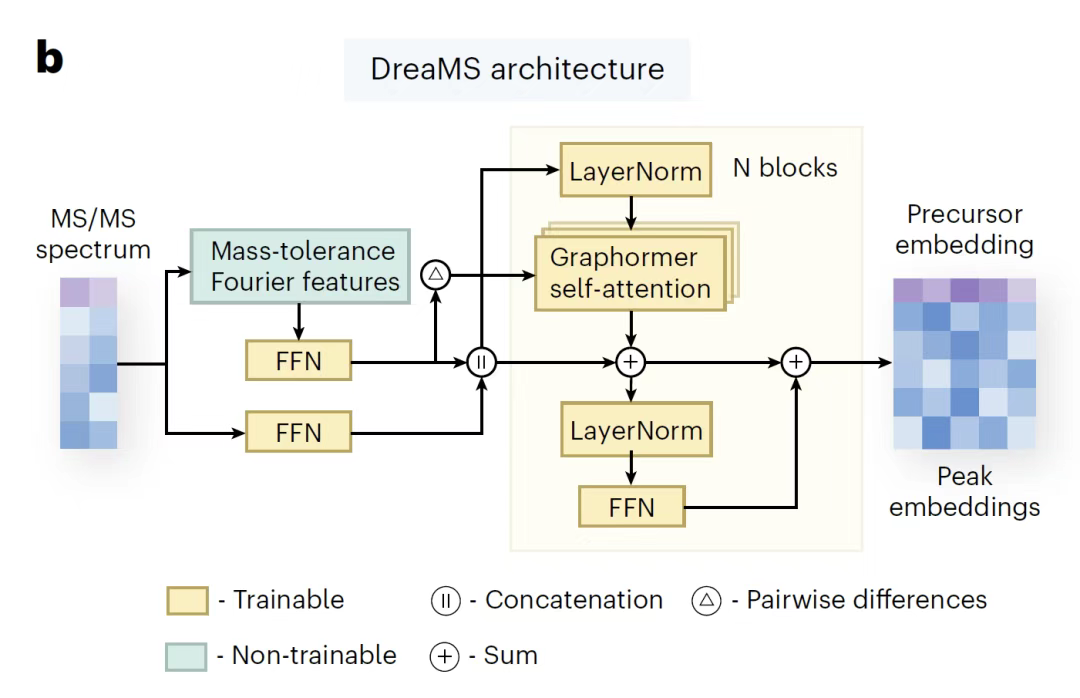
Based on the GeMS dataset, the DreaMS model aims to extract molecular representations from unannotated MS/MS spectra via self-supervised learning.This model draws on the BERT architecture in natural language processing and pioneered a self-supervised learning paradigm in the field of small molecule mass spectrometry.Its core design includes two training objectives: one is to randomly mask the mass-to-charge ratio (m/z) of 30% in the spectrum in proportion to the intensity, and train the model to reconstruct the masked peaks, while introducing "parent ion tags" to aggregate spectral-level information (similar to the sentence-level representation of language models); the other is to learn to predict the chromatographic elution order through spectral pairs of the same LC-MS/MS experiment, and strengthen the intrinsic relationship between molecular structure and peak elution rules.
In terms of model architecture, as shown in the figure below,DreaMS is based on a 7-layer Transformer encoder equipped with an 8-head self-attention mechanism, which can generate a 1,024-dimensional representation vector.For high-resolution mass-to-charge ratio data, the model uses Fourier features preprocessing technology to decompose continuous mass values into sine/cosine frequency components, capture the details of integer and floating-point parts, and further associate element composition predictions through a feedforward network; the intensity value is processed by a shallow network and concatenated with the Fourier features as Transformer input. In addition,DreaMS explicitly introduces the Fourier feature differences of all peak pairs into the self-attention head (borrowing from the Graphormer architecture).Directly model the neutral loss relationship, avoiding additional labeling or complex calculations.
This study used a linear probing technique to evaluate changes in representations acquired during the training phase.First, during the training process, the logistic regression model based on the parent ion embedding vector can gradually predict the MACCS bond fingerprint, indicating that the model learns molecular fragment information in self-supervision; second, the attention head analysis shows that the model prioritizes the characteristic peaks representing the molecular structure rather than the noise; finally, the characterization space clustering results show that even the spectra under different ionization conditions can be linearly distributed according to the molecular structure, verifying its ability to capture structural features.
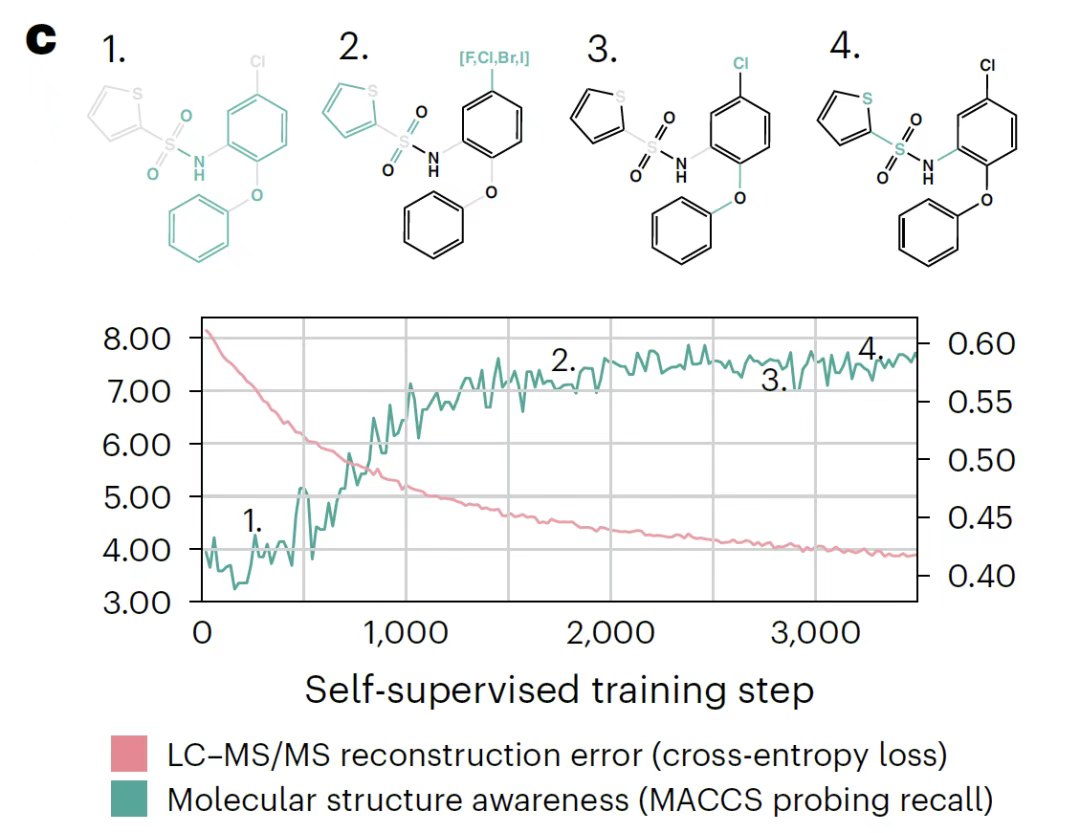
DreaMS model cross-task migration: mass spectrometry analysis from single-molecule analysis to whole metabolome interconnection
As the first mass spectrometry analysis model based on self-supervised learning, the DreaMS model has shown significant advantages in cross-task migration capabilities. The research team adapted it to four core tasks:
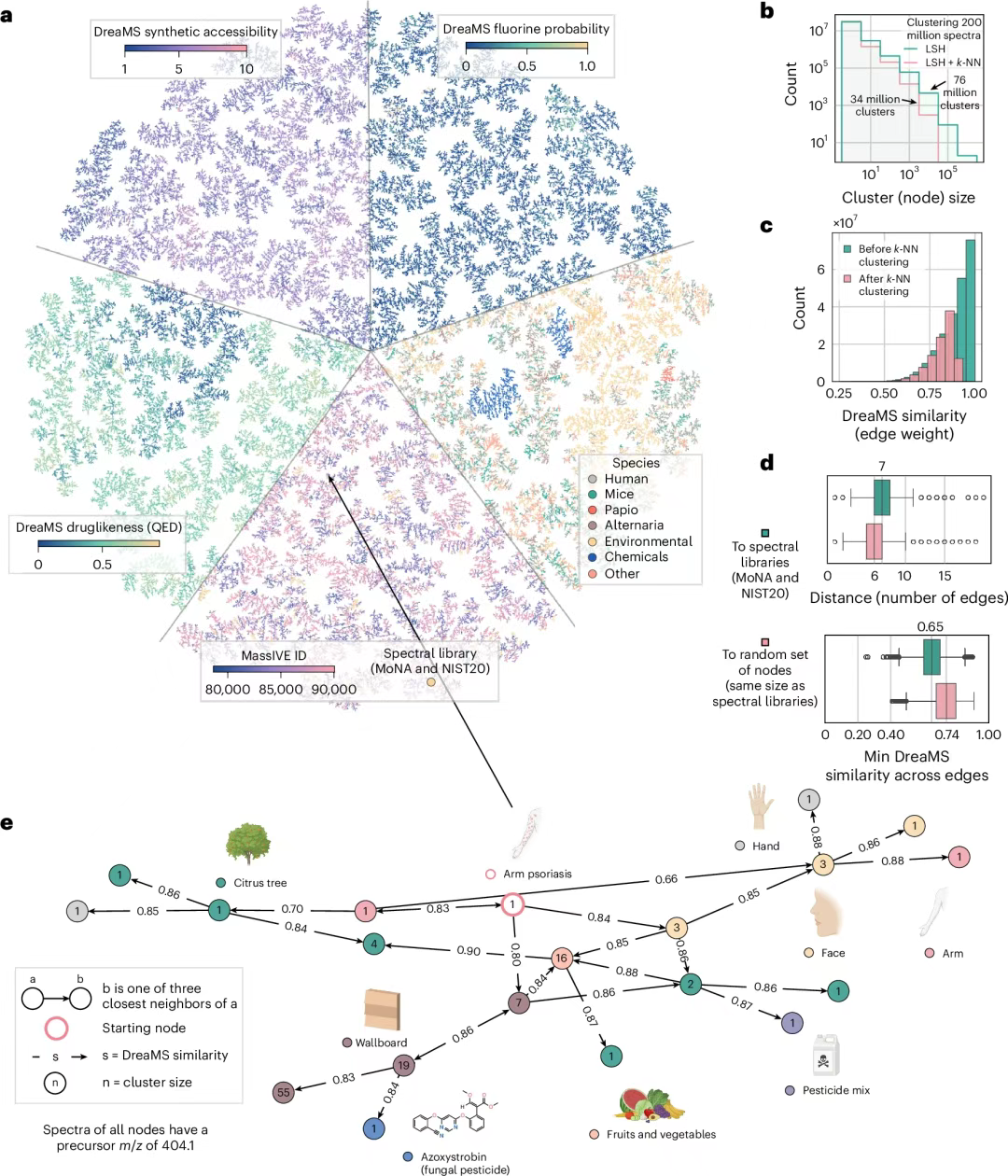
In spectral similarity analysis,As shown in the figure below, the model first achieves zero-sample matching through self-supervised characterization. The correlation between the cosine similarity of the embedding space and the similarity of the molecular structure (such as the Tanimoto coefficient) exceeds the supervised algorithm MS2DeepScore that relies on labeled data training. In view of the limitation that zero-samples are insensitive to subtle differences in molecular structure, a triplet of difficult examples including reference spectra, positive samples of the same molecule, and negative samples of similar mass are designed for comparison and fine-tuning, so that in the retrieval task with a precursor mass deviation within 10ppm,The fine-tuned DreaMS significantly outperforms 44 traditional similarity metrics.Moreover, the embedding results are more robust to differences in mass spectrometry instruments, and UMAP analysis shows that its representation space is strictly clustered according to molecular chemical formulas and structural motifs.
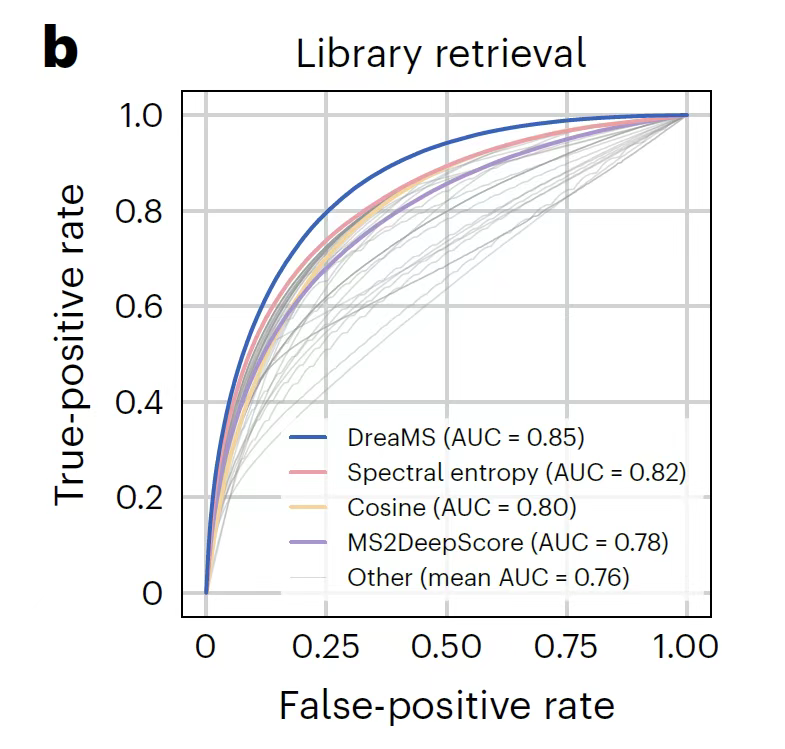
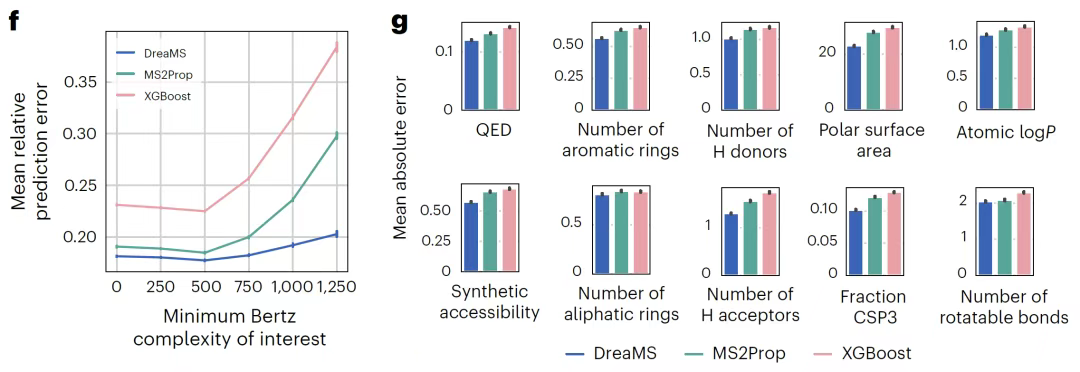
In the molecular fingerprint prediction task,As shown in the figure below, DreaMS breaks through the complex process of traditional methods that rely on chemical formula assignment or fragment tree generation. A single forward pass can directly predict Morgan fingerprints from raw spectra. The performance of searching the PubChem database is comparable to that of the deep learning model MIST that relies on peak chemical formula annotation, but it omits the intermediate computationally intensive steps. For the prediction of pharmaceutical-related chemical properties, the model outputs Lipinski's five rule parameters, Bertz molecular complexity and other indicators through fine-tuning.It has achieved the current best performance in both large-scale drug screening and extraterrestrial biomarker search scenarios.
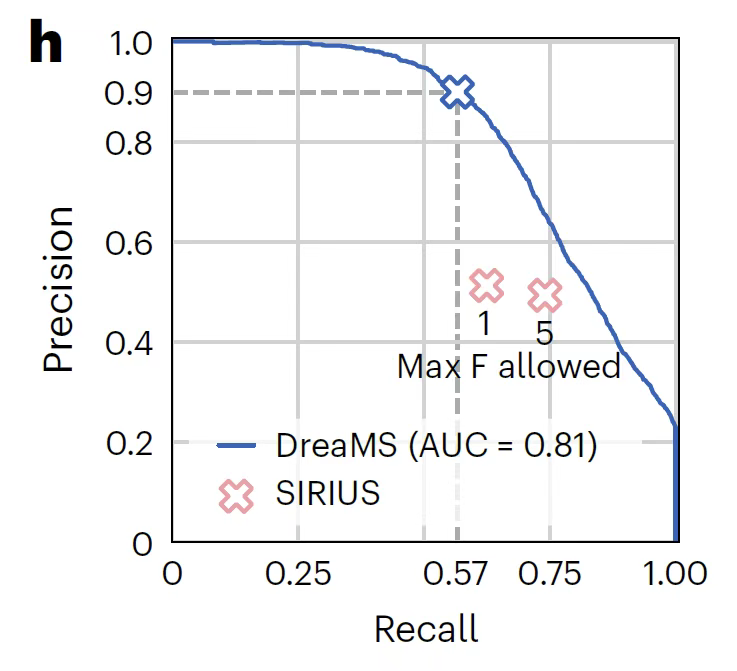
In the most challenging task of detecting fluorinated molecules,As shown in the figure below, DreaMS achieves a precision of 0.91 and a recall of 0.57 through a probabilistic prediction model.This is far superior to the SIRIUS algorithm which relies on fragmentation rule combination search and has an accuracy of only 0.51.In particular, it exhibits strong generalization ability in the detection of molecules with novel structures, providing a key tool for fluoride-related drug development and environmental monitoring.
Based on high computational efficiency (the calculation of embedding 1 million spectra takes only 1 hour on the NVIDIA A100 GPU), as shown in Figure ad below, the research team constructed a DreaMS graph containing 201 million mass spectra, and generated a three-nearest neighbor (3-NN) graph of 34 million nodes through local sensitive hashing clustering. The edge similarity of 67% is higher than 0.8, and 99.7% nodes form a single connected component. The shortest path analysis shows that it can connect any spectrum with a known library entry within 6 steps.
In a metabolomics study of arm psoriasis,As shown in Figure e below, the map reveals the potential association between the disease and the fungicide pyraclostrobin through spectral connectivity. The association pathway involves environmental exposure sources such as contaminated food and treated trees, providing a new data-driven perspective for the exploration of the causes of complex diseases. This ability to accurately annotate a single task to infer the entire library network marks a new era in which mass spectrometry analysis technology has moved from "single molecule decoding" to "entire metabolome interconnection."
Industry-university-research collaboration drives innovation in mass spectrometry technology
In the field of small molecule mass spectrometry analysis and metabolomics research, universities and companies around the world are using innovative technologies to promote breakthroughs in this field.
In terms of university research, AI-assisted multi-omics big data analysis technology developed by Hu Zeping's laboratory at Tsinghua University in China, combined with high-precision metabolomics methods, successfully revealed the metabolic interaction mechanism between neurons and cancer cells in the tumor microenvironment and discovered neurotransmitter regulatory pathways that can be used as therapeutic targets. Its results have been reviewed by Nature journals many times. The "CataAI Characterization Expert System" developed by the Dalian Institute of Chemical Physics of the Chinese Academy of Sciences,By integrating deep learning technology into the mass spectrometry data analysis process and using self-built databases and new algorithms, we have achieved intelligent recommendations from mass spectra to molecular structures.A two-stage neural network model was developed for the complex characterization data of energy catalytic materials.
The Global Natural Products Social Molecular Network (GNPS) platform of the University of California, San Diego (UCSD)As the source of the core dataset GeMS of the DreaMS model studied in this article, it continues to promote cross-institutional mass spectrometry data sharing and integration.Its latest research established a high-throughput intestinal microbiome metabolomics analysis method by comparing ethanol and methanol solvent systems, providing a standardized process for analyzing host-microbe interaction mechanisms.
In corporate innovation practices, the American company Agilent has launched a new generation of liquid quality detection systems such as the Pro iQ series, which have excellent performance and sensitivity and are ideal for complex biological molecule monitoring and impurity detection.Its mass range is extended to m/z 2–3000 and its sensitivity is enhanced by Agilent Jet Stream (AJS) technology.It supports routine and trace detection of small molecules and macromolecules, providing disruptive technical means for food safety supervision. Based on liquid chromatography-tandem mass spectrometry technology, the Chinese company Kailaipu Technology has independently developed more than 20 clinical mass spectrometry kits, covering more than 300 detection items, among which the detection reagents for catecholamine metabolites in blood and urine have been included in the expert consensus of the Chinese Medical Association Endocrinology Society and become the clinical gold standard.
In general, the current field of small molecule mass spectrometry analysis and metabolomics research is undergoing a technological innovation led by universities and enterprises. These innovations not only deepen human understanding of the complexity of biological systems in theory, but also show great potential in practical applications, from early cancer diagnosis to cardiovascular disease prognosis prediction, from catalytic material research and development to food safety supervision. This revolution triggered by the resonance of algorithm innovation and experimental science may completely reconstruct the entire chain ecology from basic research to clinical application, bringing more far-reaching impacts to related fields.
Finally, I would like to recommend an event to everyone. HyperAI will hold the 7th Meet AI Compiler Technology Salon in Beijing on July 5.We are fortunate to have invited many senior experts from AMD, Peking University, Muxi Integrated Circuit, etc. Welcome everyone to click the link below to sign up~
https://www.huodongxing.com/event/1810501012111
Reference articles:
1.https://mp.weixin.qq.com/s/1QUjLMtj_6ui9T0gbuZtrA
2.https://dicp.cas.cn/xwdt/ttxw/202411/t20241107_7435521.html
3.https://ccms-ucsd.github.io/GNPSDocumentation/
4.https://mp.weixin.qq.com/s/Wgh2w0G76koqc9AY0PBHcg








