Command Palette
Search for a command to run...
Published in Nature Sub-journal! Peking University Team Uses AI to Predict the Evolution Direction of COVID-19/AIDS/influenza Viruses, With an Accuracy Improvement of 67%
In December 2019, the COVID-19 outbreak suddenly occurred. This highly contagious disease caused by the SARS-CoV-2 virus, in just one month, the number of cases in my country exceeded 1,000 and quickly spread to the world.
In order to combat the further spread of the virus, our country launched a universal free vaccination policy in early 2021. However, even with the support of vaccines, the health crisis has become increasingly complicated.This is because the SARS-CoV-2 virus continues to mutate.Gradually adapting to the immune pressure generated by the vaccine and the changing environmental conditions, the "virus strain" originally discovered in Wuhan has long disappeared, and has been replaced by various mutant strains, which continue to trigger a new wave of infection, and its impact will continue until after 2023.
Coincidentally, just recently, the influenza virus positivity rate has continued to rise, and many people have been infected with influenza A (H1N1) without knowing it. Similar to the SARS-CoV-2 virus, H1N1 is also highly contagious, spreads rapidly, and mutates rapidly. Multiple subtypes of the virus may appear in the same season, which also increases the risk of repeated infection in the population within a short period of time.
This shows that predicting the direction of virus evolution is crucial for prevention and control and vaccine and drug design.However, mutations are the basis of viral evolution, and their high randomness means that usually only a very small number of mutations can "just" increase the adaptability of the virus. This imbalance between positive samples (beneficial mutations) and negative samples (harmful mutations) makes it extremely difficult to train a deep learning model that can predict rare beneficial mutations of the virus. At the same time, viruses often only mutate at a few sites, which makes it difficult for neural networks to directly capture the weak changes in intramolecular interactions caused by mutations, and also causes trouble for modeling.
In this regard, Professor Tian Yonghong and Associate Professor Chen Jie from the School of Information Engineering at Peking University, together with Researcher Zhou Peng from the Guangzhou National Laboratory, guided doctoral student Nie Zhiwei and master student Liu Xudong to re-examine the problem of predicting viral evolution and proposed an evolution-driven virus mutation driving force prediction framework E2VD.This framework can predict the evolutionary direction of SARS-CoV-2 virus, Influenza virus, Zika virus and HIV (AIDS virus), significantly improving the speed of human response to emerging viral infections and providing important support for the rapid optimization of vaccines and drugs.
The research was published in Nature Machine Intelligence on January 17, 2025 under the title "A unified evolution-driven deep learning framework for virus variation driver prediction".

Paper address:
https://www.nature.com/articles/s42256-024-00966-9
Paper address: Follow the official account and reply "Viral Evolution" to get the full PDF
The open source project "awesome-ai4s" brings together more than 100 AI4S paper interpretations and provides massive data sets and tools:
https://github.com/hyperai/awesome-ai4s
Dataset: UniRef90 pre-training dataset and virus deep mutation scanning dataset
Viruses will continuously generate new mutations and selectively accumulate them during their evolution. Therefore, protein language models for evolutionary scenarios need to have strong zero-shot generalization capabilities, that is, they need to be able to handle unseen mutations. To achieve this,The research team chose UniRef90 as the dataset for pre-training of the protein language model. UniRef90 contains rich sequence-level evolutionary information without negatively affecting performance in the early stages of model training. This rich evolutionary information allows the model to be exposed to enough protein family sequence samples during pre-training, thereby improving its zero-shot generalization ability.
In addition, to support the model learning the evolutionary fitness landscape caused by viral mutations,The research team used open source deep mutation scanning datasets of various viruses.
Model Architecture: Evolution-Inspired Universal Architecture Design
Based on the design of "weak mutation amplification" and "rare beneficial mutation mining", the research team proposed an evolution-driven virus mutation driving force prediction framework E2VD. As shown in Figure a below,It mainly includes 3 modules:They are protein sequence encoding, local-global dependence coupling, and multi-task focal learning.
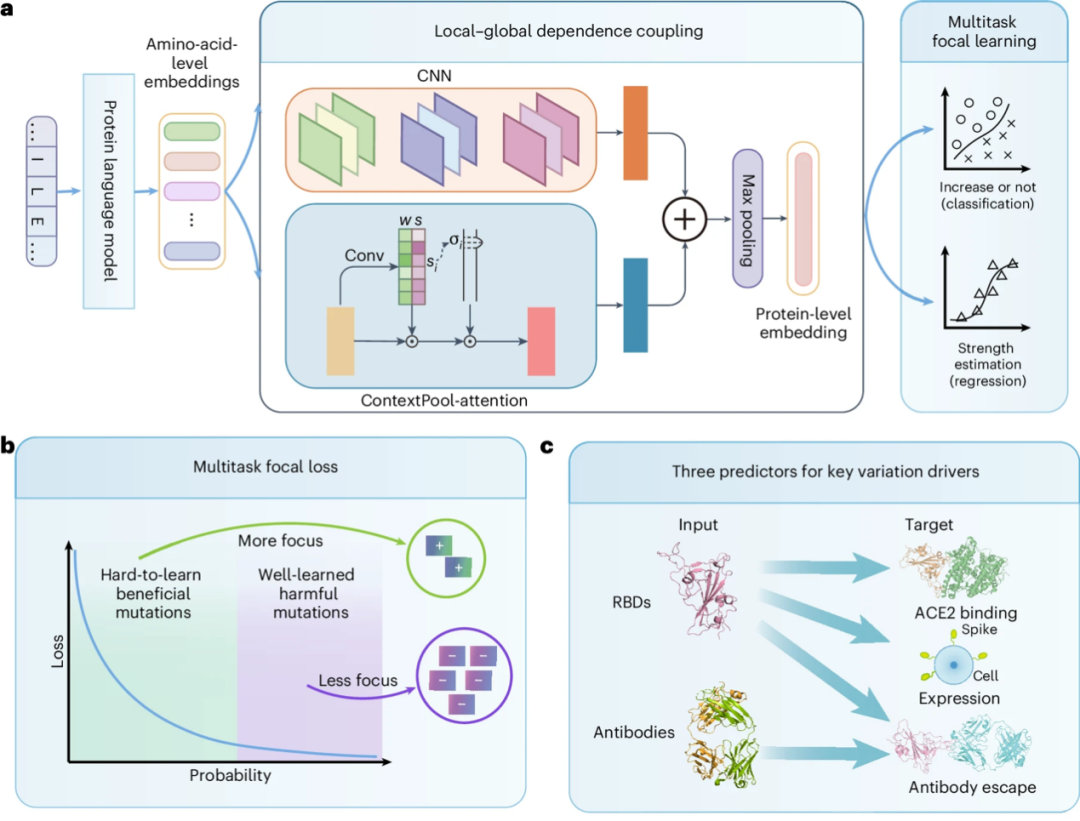
* first,In the protein sequence coding module, the research team independently trained a customized protein language model for viral evolution, which can accurately extract the characteristics of viral protein sequences;
* Secondly,In the local-global interaction dependency fusion module, researchers used convolutional neural networks (CNNs) to capture the interaction dependency between mutations and adjacent amino acids, and designed a learnable dynamic attention mechanism to construct a long-range interaction dependency network at the motif level where the mutation is located. This design effectively solves the problem that weak effects caused by fewer overall mutations in the variant are difficult to capture;
* Then,In the multi-task focus learning module, the advantages of multi-task learning and difficult sample mining strategies are combined to improve the model's predictive performance for virus mutation fitness through parameter sharing of multi-task training.
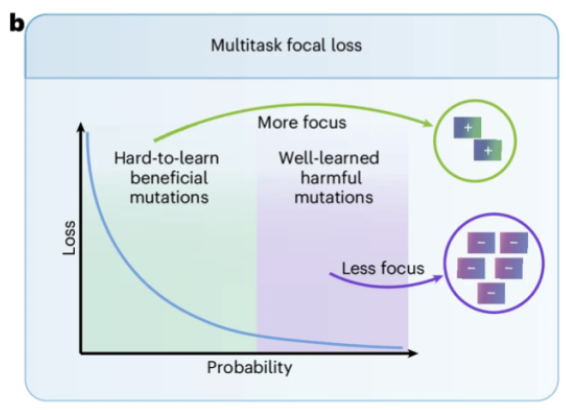
More importantly, as shown in Figure b above, the team designed a novel multi-task focal loss function, which prompts the model to pay more attention to rare beneficial mutations that are difficult to learn effectively during training, thereby greatly improving the prediction performance for rare beneficial mutations (i.e., difficult samples).
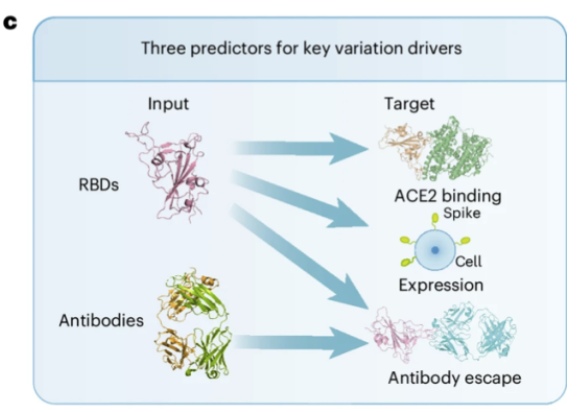
In addition, as shown in Figure c above, the E2VD prediction framework can flexibly adjust the input and output for various virus fitness prediction tasks. For example, to predict changes in binding affinity caused by mutations, only the virus sequence can be input; to predict changes in antibody escape ability caused by mutations, both the virus sequence and the antibody sequence can be input, etc., thereby achieving high-precision evolutionary predictions across virus types and strains on a unified architecture.
Specifically, in the study, the E2VD framework was used for prediction tasks related to SARS-CoV-2 virus, Influenza (influenza virus), Zika (Zika virus) and HIV (AIDS virus):
* Tasks for SARS-CoV-2 include prediction of binding affinity, expression, and antibody escape, which are the key drivers of virus mutation.
* The task for Influenza, Zika and HIV viruses is to predict the fitness effect caused by mutations in order to analyze the generalization ability of the model.
Experimental results: E2VD improves the accuracy of predicting beneficial mutations by 67%, and has excellent generalization performance
E2VD can accurately capture viral evolution patterns and improve the accuracy of predicting beneficial mutations by 67%
The team compared the prediction performance of the customized protein language for evolutionary scenarios with that of the mainstream protein language model. The results showed that the team's customized protein language model achieved the best prediction performance with a minimum of 340M model parameters, even surpassing the ESM2-15B, which has 44 times the number of parameters. This proves the effectiveness of the customized pre-training dataset and training strategy.
Subsequently, the team compared E2VD with mainstream methods in various key virus evolution driving force prediction tasks. The results showed that E2VD significantly outperformed other methods, with performance improvements ranging from 7% to 21%. Furthermore, in order to prove the ability of E2VD to accurately capture virus evolution patterns, such as accurately distinguishing different types of mutations and accurately mining rare beneficial mutations, the researchers conducted multiple experiments.
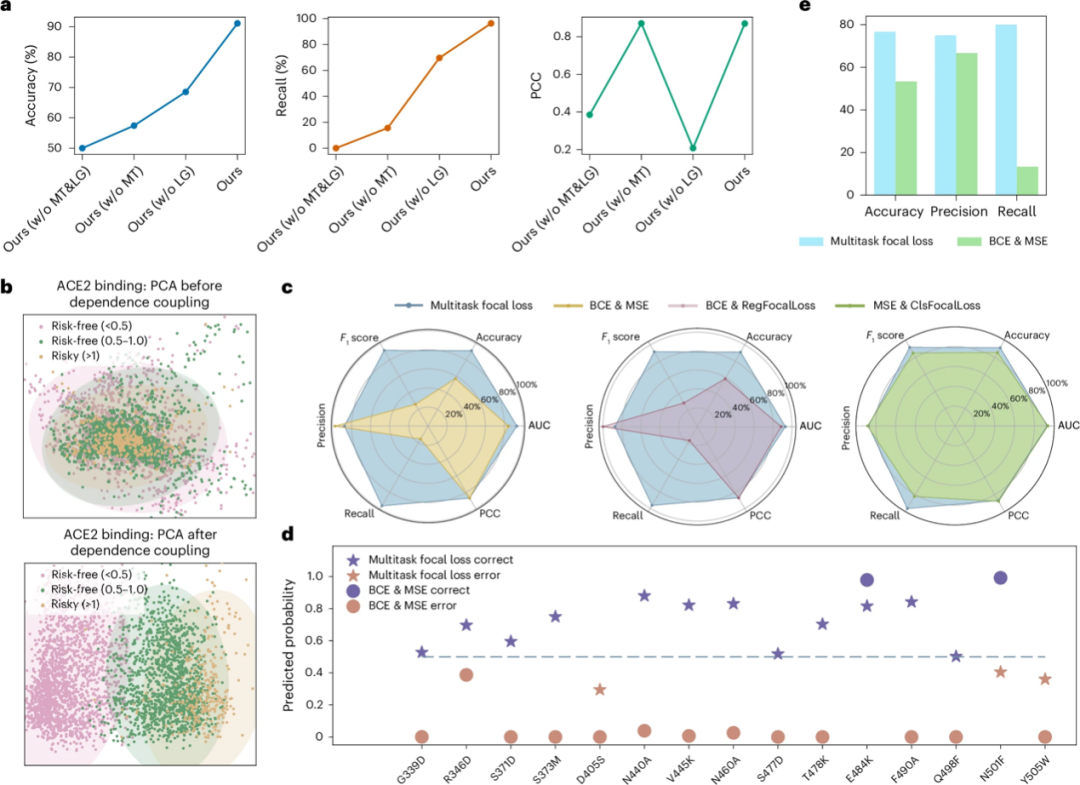
a: w/o MT means E2VD without MT module; w/o LG means E2VD without LG module; w/o MT&LG means E2VD without MT&LG module
b: Three mutation types with risk levels described in the binding affinity prediction task
d: The ability of different losses to capture rare beneficial mutations
First, a module ablation study was performed to explore the contribution of the local-global interaction dependency fusion (LG) module and the multi-task focus learning (MT) module to the prediction performance. As shown in Figure a above, the study found that the MT module was effective in mining scarce beneficial mutations in viral fitness (the recall rate increased from 0 to 69.63%). Combining the LG module with the MT module can further improve the model performance, with an accuracy of 91.11%, a recall of 96.3%, and a correlation coefficient of 0.87.
The multi-task focal loss function proposed by the team can significantly improve the prediction performance. In order to evaluate the ability of multi-task focal loss in capturing rare beneficial mutations, the researchers selected representative beneficial mutations and harmful mutations to form a test set.
* In terms of prediction of beneficial mutations, as shown in Figure d above, E2VD improves the prediction accuracy of rare beneficial mutations from 13% to 80%, achieving a leapfrog improvement in accuracy, thereby accurately and efficiently mining rare beneficial mutations that are crucial to viral evolution.
* For harmful mutations, multi-task focal loss and traditional BCE&MSE perform similarly. This is because BCE&MSE cannot help the model learn the scarce beneficial mutations, which makes the model tend to predict all mutations as harmful mutations.
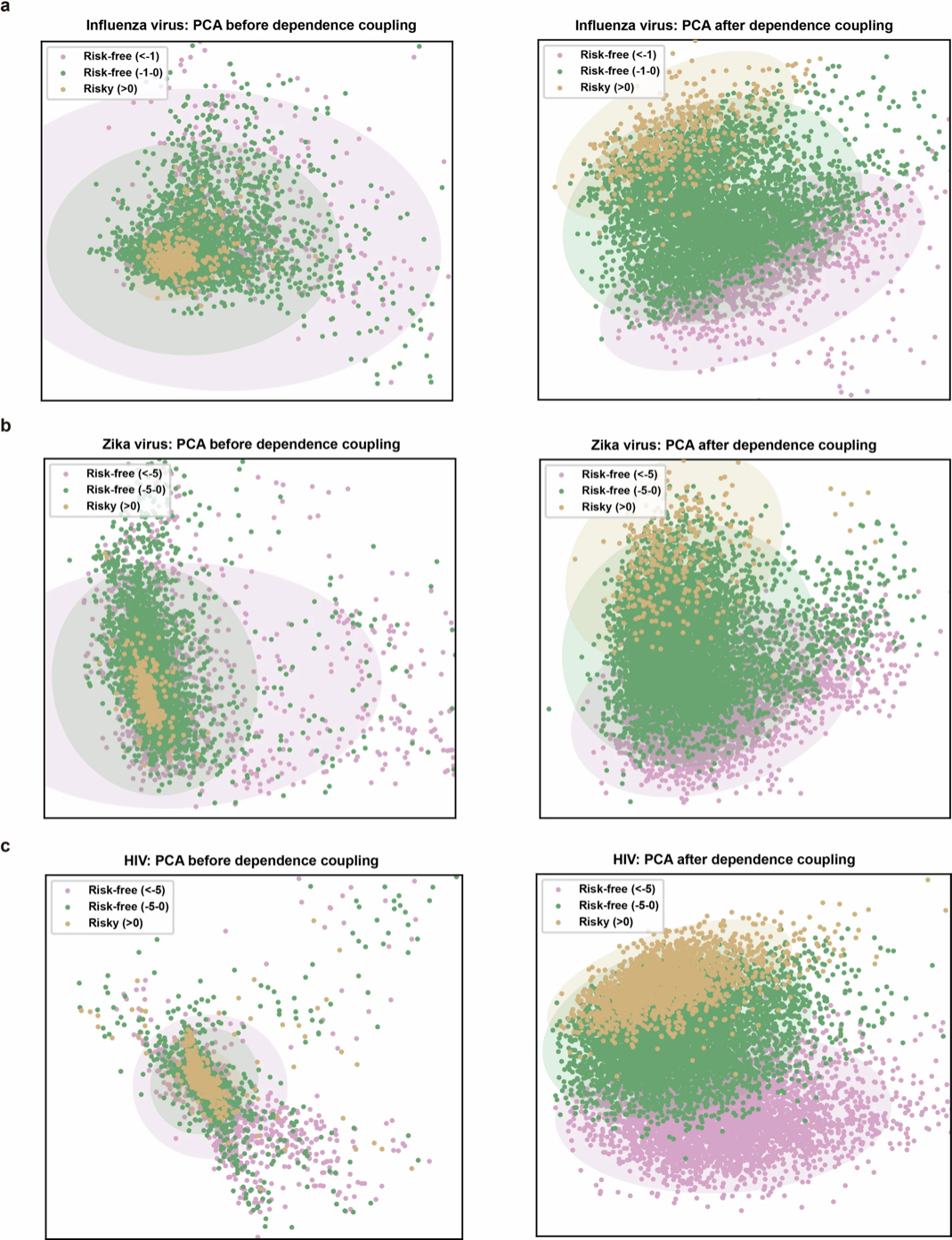
As shown in Figure b below, the researchers used principal component analysis (PCA) to visualize the dimensionality reduction of three types of mutations in influenza, Zika and HIV. The results showed that after being processed by the LG module, the characteristics of different mutations were clearly distinguished and the boundaries were clear. This shows that LG can enhance the sensitivity of E2VD to various types of mutations by capturing and reconstructing the intramolecular interaction network, thereby better understanding the evolutionary adaptability of the virus.
E2VD has excellent generalization performance and can make predictions across virus types and strains
Viruses continue to evolve under selection pressure, leading to the emergence of multiple strains. For example, the influenza virus that has attracted much attention recently includes multiple types and shows seasonal variations. Therefore, the generalization ability of the model is crucial to cope with complex virus evolution trends. The researchers proposed the "Ordinal Pair Proportion" (OPP) to evaluate the generalization ability of the model in the prediction tasks of different strains and types of the same virus.
* OPP represents the proportion of correctly predicted mutation pairs among all mutation pairs. The larger the OPP value, the less chaotic the predicted adaptive landscape is, indicating that the model is more capable of predicting the relative order of viral mutation drivers.
As shown in Figure b below, for the cross-strain binding affinity prediction task, the researchers evaluated the OPP of 6 different strains and all strains mixed data (All), and found that E2VD significantly outperformed other methods in all cases. As shown in Figure c below, in the expression level prediction task, E2VD also outperformed other methods on the vast majority of strains. Overall, E2VD comprehensively surpassed the most advanced methods on out-of-distribution strains and showed highly generalized performance.
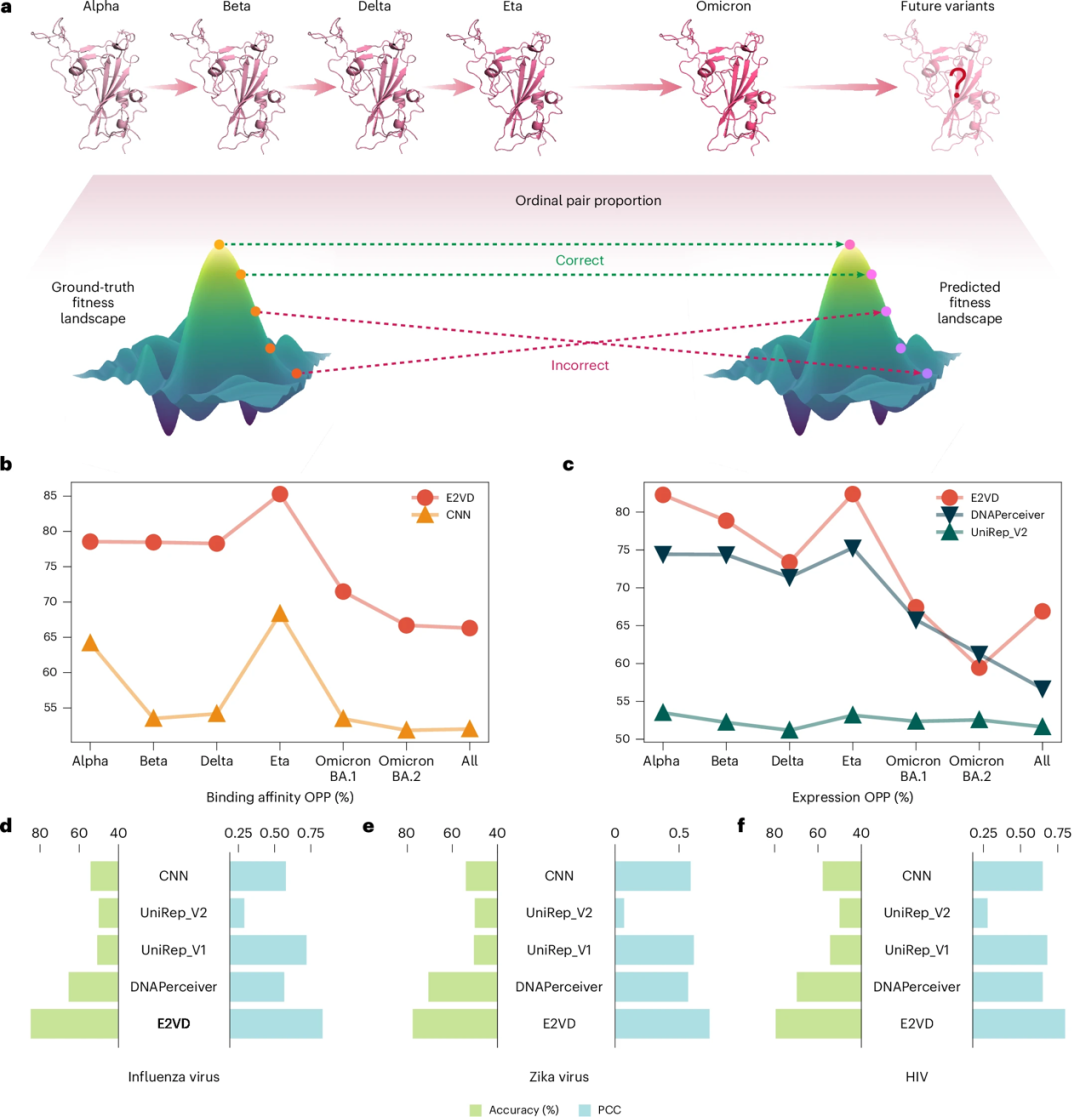
* b, c: E2VD predicts the OPP of different virus strains; d, e, f: E2VD predicts the performance of different types of viruses
As shown in Figures d, e, and f above, in cross-virus type predictions, the researchers found that E2VD exhibited ideal generalization capabilities for the new coronavirus, Zika virus, influenza virus, and HIV, surpassing other methods in all aspects, and may be further expanded to more infectious viruses in the future.
AI has great potential in predicting viral evolution
The above research re-explored the problem of virus evolution prediction from the perspective of evolution, and constructed a universal evolution prediction framework E2VD suitable for different virus types and strains. This framework showed excellent predictive performance and generalization ability in multiple virus mutation driver factor prediction tasks, making it possible to predict virus evolution trends.Furthermore, the flexible and customized combination of E2VD can also realize the prediction of evolutionary trends at different scales.
* First, E2VD can explain the path of viral evolution during pandemics, helping us understand the reasons for the prevalence of strains and the molecular mechanisms behind them.
* Secondly, combined with virtual deep mutation scanning simulation, E2VD is able to predict possible high-risk mutations, achieving a hit rate of 80%.
* Finally, E2VD also achieves pandemic-scale macroevolutionary trajectory prediction, reproducing the evolutionary path of the virus in the real world, thus providing theoretical support for the interpretation of the virus evolution mechanism.
In the future, the team plans to combine E2VD with vaccine and protein drug design processes to improve the efficiency and controllability of the design, which will be of great significance to viral prevention and control and drug design.
It is worth mentioning that the authors of the study are Professor Tian Yonghong and Associate Professor Chen Jie from the School of Information Engineering of Peking University, and their doctoral students Nie Zhiwei and Master's student Liu Xudong. The team continues to focus on research in the field of AI for Life Science. Their project "Ahead of the Evolution of the Virus - Predicting Future High-Risk Coronavirus Mutants through Artificial Intelligence Simulation" was successfully shortlisted for the 2022 "Gordon Bell New Crown Special Award" in November 2022 (the Gordon Bell Prize is the highest academic award in the field of high-performance computing applications in the world).
The team has profound experience in the field of virus evolution prediction. In July 2023, the team published "Running ahead of evolution—AI-based simulation for predicting future high-risk SARS-CoV-2 variants" in The International Journal of High Performance Computing Applications. Specifically, the researchers pre-trained a large protein language model and constructed a high-throughput screening method based on binding affinity and antibody escape predictions. This is the first study on the simulation of SARS-CoV-2 RBD mutations. The model successfully identified mutations in the RBD region of 5 variants of concern and screened out millions of potential variants in seconds, providing a technical means for epidemic prevention and control in an "AI+HPC" (artificial intelligence + high-performance computing) paradigm.
Paper link:
https://journals.sagepub.com/doi/abs/10.1177/10943420231188077
In addition, the team has developed a series of basic models for life sciences. Taking the "enzyme-substrate" interaction prediction task, which is crucial to enzyme engineering, as an example, the team released a preprint article in December 2024, proposing a progressive conditional deep learning framework MESI for multi-purpose enzyme-substrate interaction prediction.
Paper link:
https://www.researchsquare.com/article/rs-5516445/v1
Specifically, by decoupling the modeling of enzyme-substrate interactions into a two-stage learning process, two conditional networks are designed to introduce enzyme reaction specificity and key catalytic interaction information, respectively, thereby promoting the gradual transition of the feature latent space from the general domain of proteins and small molecules to the catalysis-aware domain. The model consistently outperforms state-of-the-art methods in various downstream tasks. In addition, the proposed conditional network implicitly captures the basic modes of enzyme catalysis, while the additional computational overhead is negligible. Supported by this conditional-aware mechanism, the model can accurately identify active sites and mine enzyme residues and substrate functional groups involved in key catalytic interactions in an efficient and low-cost manner without the need for any structural information.
With the assistance of artificial intelligence, the team will further promote in-depth research in related fields of AI for life science, open up more possibilities for virus prediction, protein drug design, vaccine development, etc. We look forward to more of their achievements.
References:
https://www.who.int/
https://news.pku.edu.cn/jxky/90d276ae5f8441849fd04372fd872154.htm
https://news.pkusz.edu.cn/info/1003/8711.htm









