Command Palette
Search for a command to run...
Hundert Universitäten Haben Die Weltweit Größte Multikohorten-Proteogenomikstudie Gestartet, Um Krankheitsverursachende Gene Zu Entschlüsseln Und Bestehende Medikamente Auf Der Grundlage Von Daten Von Fast 80.000 Teilnehmern Neu Zu positionieren.

Das menschliche Genom ist wie eine vollständige Bedienungsanleitung fürs Leben, die alle genetischen Informationen wie Aussehen, Größe, Körperbau und Krankheitsrisiko speichert. Diese Anleitung zu entschlüsseln ist jedoch kein einfacher Prozess; es können verschiedene „unerwartete Ereignisse“ auftreten, darunter pathogene Mutationen, die eine Veranlagung für bestimmte Krankheiten bedingen. Noch schwieriger ist es…Die meisten pathogenen Varianten befinden sich in nicht-kodierenden Regionen des Genoms, die nicht direkt Proteine kodieren.Dieser „Black-Box“-Mechanismus, der nicht angibt, welches Gen die Krankheit verursacht oder durch welchen Mechanismus, schränkt unsere Fähigkeit, die pathogenen Gene und Mechanismen zu ermitteln, stark ein. Und als die direkten Ausführenden, die Genfunktionen zum Leben erwecken,Die Tausenden von Proteinen, die im menschlichen Blut zirkulieren, sind der Schlüssel zur Entschlüsselung von Black-Box-Mechanismen und zur Verknüpfung von nicht-kodierenden Variationen mit krankheitsbezogenen Mechanismen.
Die Proteogenomikforschung hat in letzter Zeit bedeutende Fortschritte in der klinischen Pathogenese und der Identifizierung potenzieller Angriffspunkte für Medikamente erzielt, doch bestehen weiterhin Einschränkungen hinsichtlich ihrer systematischen und großflächigen Anwendung in der Humanbiologie. Erstens konzentrierte sich die bisherige Forschung fast ausschließlich auf proximale cis-wirkende Varianten (d. h. cis-Protein-Quantitative Trait Loci, cis-pQTLs).Nicht-kodierende Variationen können in regulatorischen Regionen lokalisiert sein und dadurch mehrere benachbarte kodierende Gene direkt beeinflussen.Es kann auch indirekt Proteine regulieren, die von Genen an anderen Stellen im Genom kodiert werden; zweitens ist die bisherige Forschung zur Multigen-Genstruktur von Protein-Biomarkern, die die Krankheitsdiagnose und -prognose beeinflussen, noch unzureichend; schließlich ist zur stabilen und allgemeinen Identifizierung von Protein-Quantitative-Trait-Loci eine wiederholte Validierung in verschiedenen Populationen erforderlich.Derzeit werden im Bereich der Breitband-Proteomik nur sehr wenige Validierungsstudien am Menschen dieser Art durchgeführt.
In Anbetracht dessenEin Team aus über hundert Universitäten und Forschungseinrichtungen, darunter die Queen Mary University of London und die Universität Cambridge, hat die bisher weltweit größte Multi-Kohorten-Proteogenomik-Studie veröffentlicht.Auf der Grundlage einer groß angelegten Metaanalyse von proteoglykämischen Genomen, die 38 unabhängige Forschungskohorten und insgesamt 78.664 Probanden umfasste, wurden 24.738 quantitative Merkmalsstellen von Proteinen systematisch identifiziert und mit 1.116 zirkulierenden Proteinen assoziiert, wodurch die umfangreichen genetischen regulatorischen Merkmale der Nähe und Distanz auf Proteinebene umfassend aufgezeigt wurden.
Maschinelles Lernen wurde eingesetzt, um wichtige Signalwege, Zelltypen und Gewebeursprünge, die die Konzentration zirkulierender Proteine regulieren, weiter zu analysieren und die zentrale Rolle der N-Glykosylierung im Proteinregulationsnetzwerk zu klären. Darüber hinaus ermöglicht die Unterscheidung zwischen cis- und trans-regulatorischen Unterschieden in Proteinen die effektive Aufklärung der intrinsischen Mechanismen verschiedener biologischer Phänotypen und liefert Evidenz für das Screening potenzieller Protein-Wirkstoffziele für bestimmte Krankheiten. Weiterhin lieferte die Triangulationsanalyse von trans-Stellen tiefergehende Hinweise auf „Drug Repurposing“.
Die zugehörigen Forschungsergebnisse mit dem Titel „Multi-Kohorten-Proteogenomanalysen enthüllen genetische Effekte im gesamten Proteom und Diseasome“ wurden in Cell veröffentlicht.
Forschungshighlights:
* Die bisher größte Multi-Kohorten-Proteogenomik-Studie mit 38 unabhängigen Studienkohorten und insgesamt 78.664 Teilnehmern.
* Es wurden 24.738 quantitative Merkmalsloci von Proteinen identifiziert und mit 1.116 zirkulierenden Proteinen in Verbindung gebracht, wodurch ein breites Spektrum an genetischen regulatorischen Merkmalen auf Proteinebene sowohl in unmittelbarer Nähe als auch über größere Distanzen umfassend aufgezeigt wurde.
Diese Studie klärt systematisch die Regulationsmechanismen zirkulierender Proteine auf genetischer Ebene auf und liefert damit eine wichtige theoretische Grundlage und Datenressourcen für das Verständnis der molekularen Mechanismen menschlicher Krankheiten, die Identifizierung innovativer therapeutischer Ziele und die Durchführung von Arzneimittel-Repositionierungsforschung.

Papieradresse:
https://www.cell.com/cell/fulltext/S0092-8674(26)00385-5
Größter Datensatz: 38 internationale Kohorten, fast 80.000 Teilnehmer
Diese Studie stellt die weltweit größte proteogenomische Metaanalyse mehrerer Kohorten dar.Die Studie integriert 38 internationale Kohorten mit insgesamt 78.664 Teilnehmern europäischer Abstammung.Auf der Grundlage der Analyse von 1.161 Blutprotein-Targets mittels der Olink-Hochdurchsatz-Proteomik-Technologie wurden 24.738 fein kartierte pQTLs (darunter 5.040 cis-pQTLs und 19.698 trans-pQTLs) identifiziert und genetische Regulationsdaten für 1.116 wirksame Proteine gewonnen.
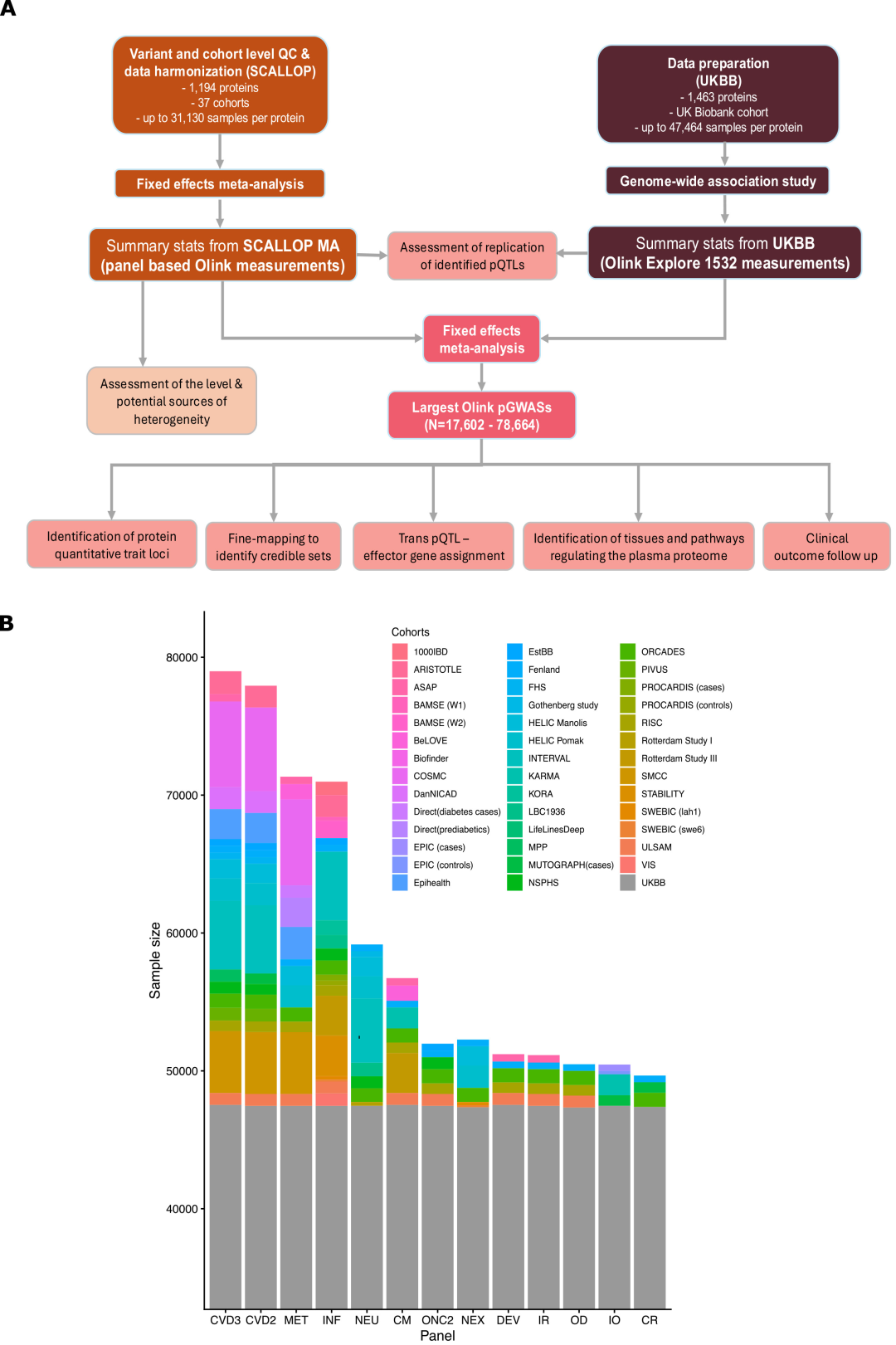
Metaanalysen zu Jakobsmuscheln:Diese Daten umfassen Genomstatistiken aus 37 Kohorten und 1.194 Blutprotein-Targets, wobei die Mehrheit der Teilnehmenden europäischer Abstammung war. Antikörperbasierte Proteomik-Assays wurden mit mindestens einem der 13 Target-96-Assay-Panels von Olink durchgeführt. Jedes Panel ist in der Lage, 92 Protein-Targets aus den Bereichen Herz-Kreislauf, Immunsystem, Entzündung, Neurologie und Stoffwechsel nachzuweisen.
Britische Biobank (UKBB):Die Studie umfasste 48.017 Teilnehmer europäischer Abstammung. Die für diese Studie verwendeten Proteomik-Messungen wurden mit der Olink Explore 1536-Plattform generiert, wobei auch antikörperbasierte Techniken zur Messung von 1.463 Proteinzielen zum Einsatz kamen.
Stufenweiser Machine-Learning-Klassifikator
Das Hauptziel dieser Studie, die Modelle des maschinellen Lernens einsetzt, ist die systematische, präzise und umfassende Zuordnung von „Effektgenen“ zu allen trans-pQTLs außerhalb der MHC-Region (Major Histocompatibility Complex). Dies löst das seit Langem bestehende Problem, Effektorgene in weit entfernten genomischen Regionen zu identifizieren, die mit Protein-QTLs (Quantitative Trait Loci) assoziiert sind, welche wiederum mit Blutproteinspiegeln in Zusammenhang stehen. Zu diesem ZweckInspiriert von der ProGeM-Architektur haben Forscher einen mehrstufigen Machine-Learning-Klassifikator entwickelt.
Zunächst zu den Quellen der Merkmale und Annotationen: Die Forscher integrierten multidimensionale biologische und genomische Annotationen für jede genetische Variante bzw. deren alternative Varianten (r² > 0,6). Die Annotationen auf Variantenebene umfassten den Abstand zwischen der Variante und dem Genom innerhalb eines 1-Mb-Basenfensters sowie die potenzielle funktionelle Auswirkung, die mithilfe des Variant Effect Prediction (VEP)-Tools abgeleitet wurde.
Gleichzeitig wurde für jedes Gen innerhalb eines 1 Mb Basenfensters eine Gen-Level-Annotation durchgeführt. Dazu gehörte die Gewinnung relevanter Hinweise auf QTL-Kolokalisationen auf Basis der GTEx v8 Proteinmenge-Genexpression, die Assoziation der Last seltener Varianten, die Verwendung des OmnipathR-Pakets Version 3.10.1 zur Überprüfung der Literatur und zur Bestimmung, ob es Ligand-Rezeptor/Protein-Komplexe gibt, die cis-Proteinen entsprechen, die von trans-Genen kodiert werden, sowie die Bestimmung, ob verwandte Gene am selben biologischen Signalweg beteiligt sind, basierend auf KEGG/REACTOME-Annotationsinformationen.
Anschließend erstellen wir den für das maschinelle Lernmodell benötigten Trainingsdatensatz, da es an allgemein gebräuchlichen Goldstandardvarianten für die Genzuordnung mangelt.Die Forscher nutzten ihr Vorwissen in Biologie und Genomik, um drei teilweise unabhängige Gruppen von „vermutlich echten Positiven (PTPs)“ zu erhalten.Um Verzerrungen zu vermeiden, wurde innerhalb jedes PTP-Sets nur ein cis-Protein beibehalten, und andere Gene innerhalb eines 1-Mb-Fensters wurden als negative Beispiele betrachtet. Dies umfasste insbesondere trans-Gene, die Ligand-Rezeptor-Paare kodieren oder hochkonfidente Proteinkomplexe mit cis-Proteinen bilden (n = 540), Sentinel-trans-pQTLs, die funktionellen Varianten zugeordnet sind (n = 1747), und trans-Gene mit einer signifikanten Anzahl seltener Varianten (n = 1049). Die Trainings- und Testdatensätze wurden anschließend im Verhältnis 7:3 basierend auf genomischen Regionen aufgeteilt, und die Ergebnisse wurden zehnmal wiederholt, um die Stabilität zu gewährleisten.
Hinsichtlich der Modellarchitektur und des Trainingsprozesses verwendet der in dieser Studie vorgestellte Modellalgorithmus den Random-Forest-Klassifikator. Durch die Eingabe von 10 Trainingsdatensätzen wird eine wiederholte 3-fache Kreuzvalidierung in Kombination mit einer Subsampling-Strategie durchgeführt, wodurch das Problem unausgewogener Datensätze während des Trainings behoben wird.Das Modelltraining wurde mit Hilfe des R-Sprach-Toolkits caret v6.0.94 durchgeführt. Anschließend wurde das jeweils beste Random-Forest-Modell in jedem Trainingsdatensatz durch eine Kappa-Score-Bewertung ausgewählt.
Anschließend wurden mithilfe von zehn Random-Forest-Klassifikatoren, die jeweils einem hypothetischen Datensatz mit echten positiven Ergebnissen entsprachen, alle Kandidaten-Effektorgene von trans-pQTL einzeln bewertet. Zunächst wurde der Median der zehn Klassifikatoren für denselben hypothetischen Datensatz mit echten positiven Ergebnissen ermittelt und anschließend die drei Sätze vorhergesagter Werte summiert. Gleichzeitig wurden bei der Erstellung des Klassifikationsmodells für jeden hypothetischen Datensatz mit echten positiven Ergebnissen die Merkmalsvariablen entfernt, die zur Definition echter positiver Proben verwendet wurden.
Letztendlich zeigten alle drei Klassifikationsmodelle eine stabile und zuverlässige Leistung, wobei die mittleren Kappa-Koeffizienten zwischen 0,54 und 0,57 lagen.
Die Entschlüsselung des Pathogenesemechanismus liefert genetische Beweise für die Arzneimittelentwicklung und die Umwidmung bereits bestehender Arzneimittel.
Diese Studie, die auf 38 internationalen Kohorten basiert und 78.664 Teilnehmer umfasst, führte eine genomische Metaanalyse mehrerer Kohorten durch, die auf 1.161 Blutproteine abzielte und systematisch die genetischen Regulationsmuster der zirkulierenden Proteinspiegel und deren Zusammenhang mit Krankheiten aufklärte.
pQTL-Identifizierung und -Charakteristika
Die Studie identifizierte 14.690 regionale Sentinel-Varianten. Durch Bayes’sches Fine-Mapping wurden 24.738 unabhängige und zuverlässige Variantensätze ermittelt, die 5.040 cis-pQTLs und 19.698 trans-pQTLs umfassten und 1.116 Proteinziele abdeckten. Darunter befanden sich cis-pQTLs im Protein an Position 87.1% und trans-pQTLs an Position 94.1%. Die cis-pQTLs an Position 82.3% und die trans-pQTLs an Position 83.3% wiesen eine hohe Konfidenz auf und enthielten 278 neu entdeckte cis-pQTLs sowie 4.013 trans-pQTLs. In der Kohorte nicht-europäischer Abstammung korrelierten die Effektstärken der identifizierten Positionen moderat mit denen der europäischen Kohorte (r = 0,6).Dies bestätigt die Robustheit der Ergebnisse über verschiedene Populationen hinweg.
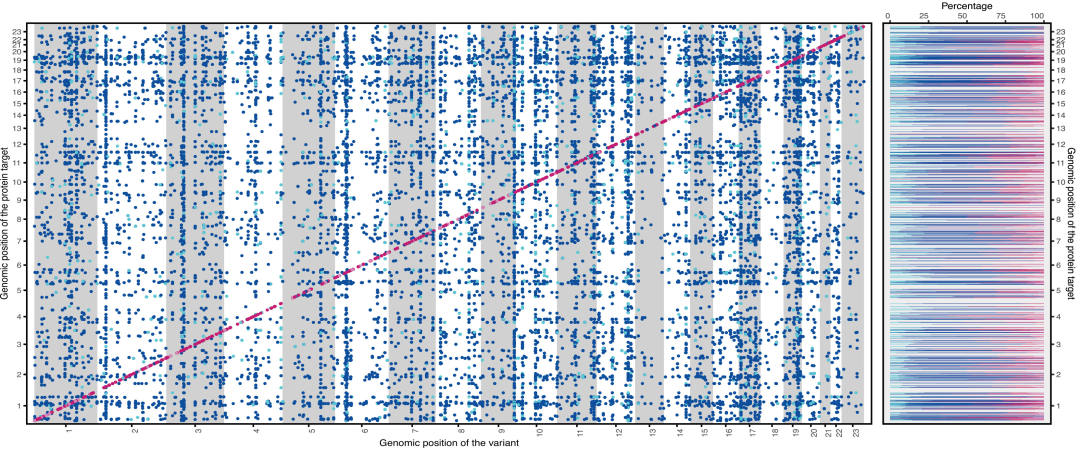
Fein lokalisierte quantitative Merkmalsloci für Proteine in SCALLOP- und UKBB-Metaanalysen
Des Weiteren bestehen signifikante Unterschiede in der Erklärungskraft genetischer Loci für Variationen der Blutproteinspiegel. Cis-pQTLs erklären durchschnittlich 8,41 TP3T der Proteinvariation, signifikant mehr als Trans-pQTLs. Proteine wie ICAM2 und FUCA1 werden jedoch hauptsächlich durch Trans-pQTLs reguliert, mit Erklärungskräften von 52,71 TP3T bzw. 68,41 TP3T, während Cis-pQTLs nur 0,31 TP3T bzw. 6,31 TP3T erklären.
Darüber hinaus ergab die weitere Beobachtung von 261 Protein-Targets keine signifikante lineare Korrelation zwischen der Erklärungskraft ihrer pQTL-Variationen und der polygenen Heritabilität, was darauf hindeutet, dass diese Studie bei der Identifizierung von pQTLs für diese Proteine eine nahezu vollständige Sättigung erreicht haben könnte.
Merkmale von Proteinzielen unter Genregulation
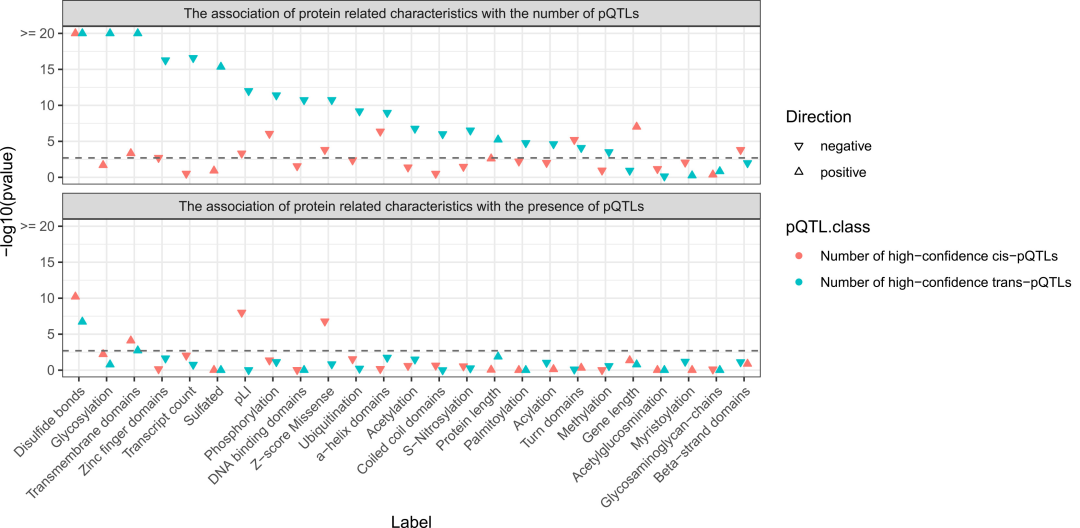
Proteineigenschaften in Bezug auf das Vorhandensein und die Menge von pQTLs basierend auf einem Zero-Inflated-Poisson-Regressionsmodell.
Proteine mit Disulfidbrücken und Transmembrandomänen weisen deutlich mehr pQTLs auf, was erklären könnte, warum diese Proteine leichter genetisch reguliert werden können; die Stärke der funktionellen Einschränkung proteinkodierender Gene korreliert hingegen signifikant negativ mit der Anzahl der cis-pQTLs.
Proteine mit einer hohen Anzahl von trans-pQTLs weisen eine signifikante Anreicherung von sekretorischen Proteinmerkmalen wie Glykosylierung und Sulfatierung auf, jedoch fehlen ihnen intrazelluläre Proteinmerkmale wie Zinkfingerstrukturen und DNA-Bindungsdomänen. Dies deutet darauf hin, dass die genetische Fernregulation zirkulierender Proteine eng mit dem Sekretionsweg zusammenhängt.
Analyse von trans-pQTL-Effektorgenen und regulatorischen Signalwegen
Auf Grundlage der Integration von biologischem Vorwissen in den Rahmen des maschinellen Lernens wurde für mehr als die Hälfte der trans-pQTLs (n = 11.261) mindestens ein Effektor-Gen mit mittlerer Konfidenz identifiziert, von denen 1.534 Zuordnungen mit hoher Konfidenz waren; für zwei Drittel der Stellen (n = 13.881) deutete die Verteilung der Kandidaten-Scores über die Gene hinweg darauf hin, dass ein einzelnes ursächliches Gen das wahrscheinlichste pathogene Gen war.
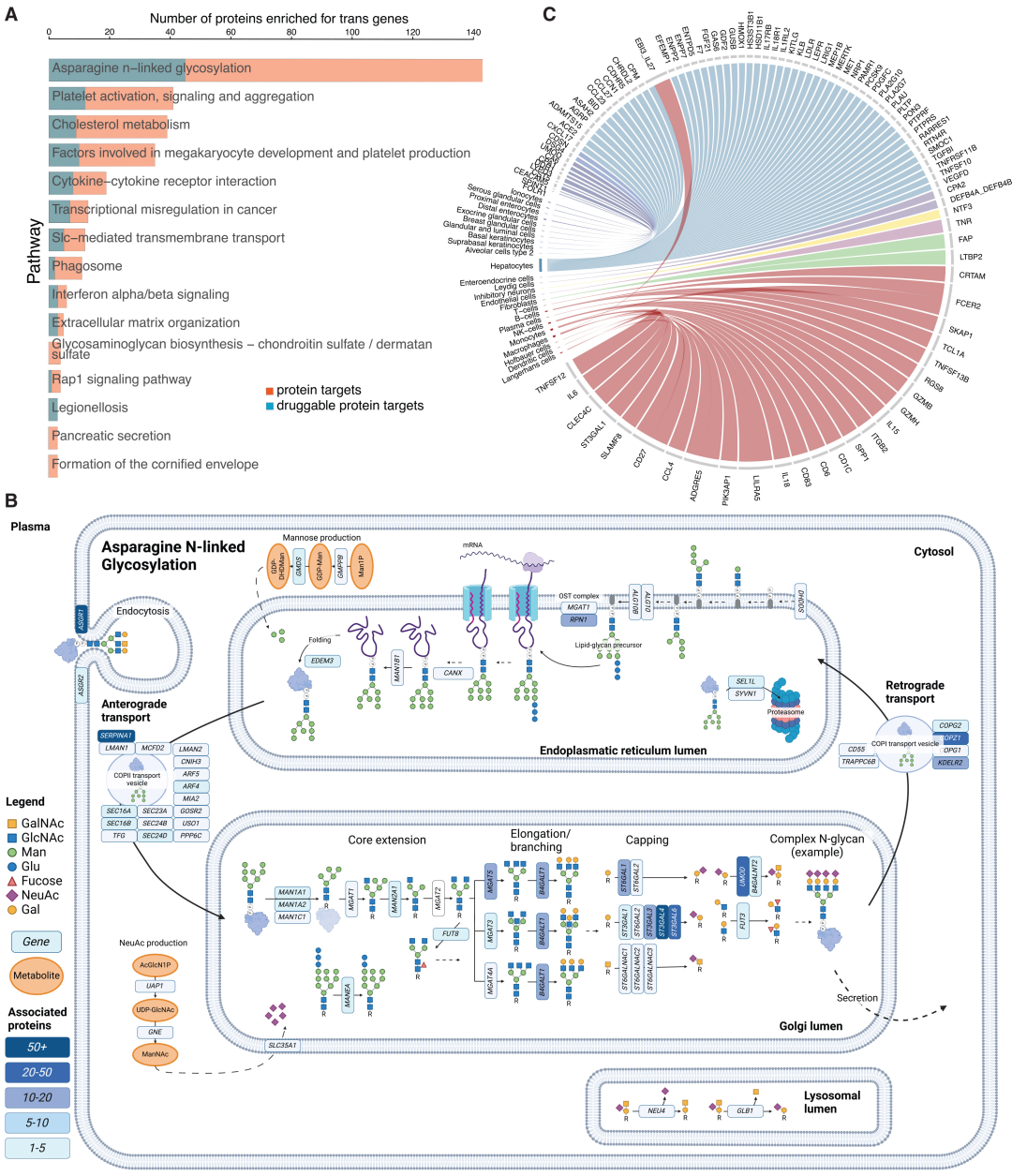
Eine Analyse der funktionellen Anreicherung ergab, dass Trans-Effekt-Gene unter anderem im Asparagin-N-Glykosylierungsweg (mit 143 Protein-Targets) und in der Thrombozytenaktivierung (mit 41 Protein-Targets) signifikant angereichert waren.Die N-Glykosylierung ist der häufigste und wichtigste regulatorische Mechanismus.
Die Ergebnisse der Zell- und Gewebeanreicherung zeigten, dass Trans-Effekt-Gene vorwiegend in Hepatozyten, natürlichen Killerzellen, Endothelzellen und Typ-II-Alveolarzellen stark exprimiert wurden. Dies deutet darauf hin, dass Leber und Immunzellen Schlüsselorte für die Fernregulation zirkulierender Proteine sind. 44 Protein-Gewebe-Paare und 76 Protein-Zelltyp-Paare waren nicht-klassisch sekretorischen Ursprungs, was die wichtige Rolle der interorganischen Kommunikation bei der Regulation der Proteinhomöostase bestätigt.
pleiotrope Effekte auf molekularer und phänotypischer Ebene
Von allen identifizierten unabhängigen pQTLs zeigten 43,41 TP3T pleiotrope Effekte, wobei trans-pQTLs signifikant höhere pleiotrope Effekte aufwiesen als cis-pQTLs. In nachfolgenden Studien wurden pleiotrope genetische Variationen in drei Typen kategorisiert: „molekular pleiotrop“, „phänotypisch pleiotrop“ und „unspezifisch pleiotrop“. Mehr als die Hälfte (332 von 533) zeigten phänotypische pleiotrope Effekte.Insbesondere wurde seine Expression in Hepatozyten um das Zweifache erhöht, und es regulierte das Zielprotein bevorzugt über Proteinkomplexe, Ligand-Rezeptor-Interaktionen und Signalwegssynergien.
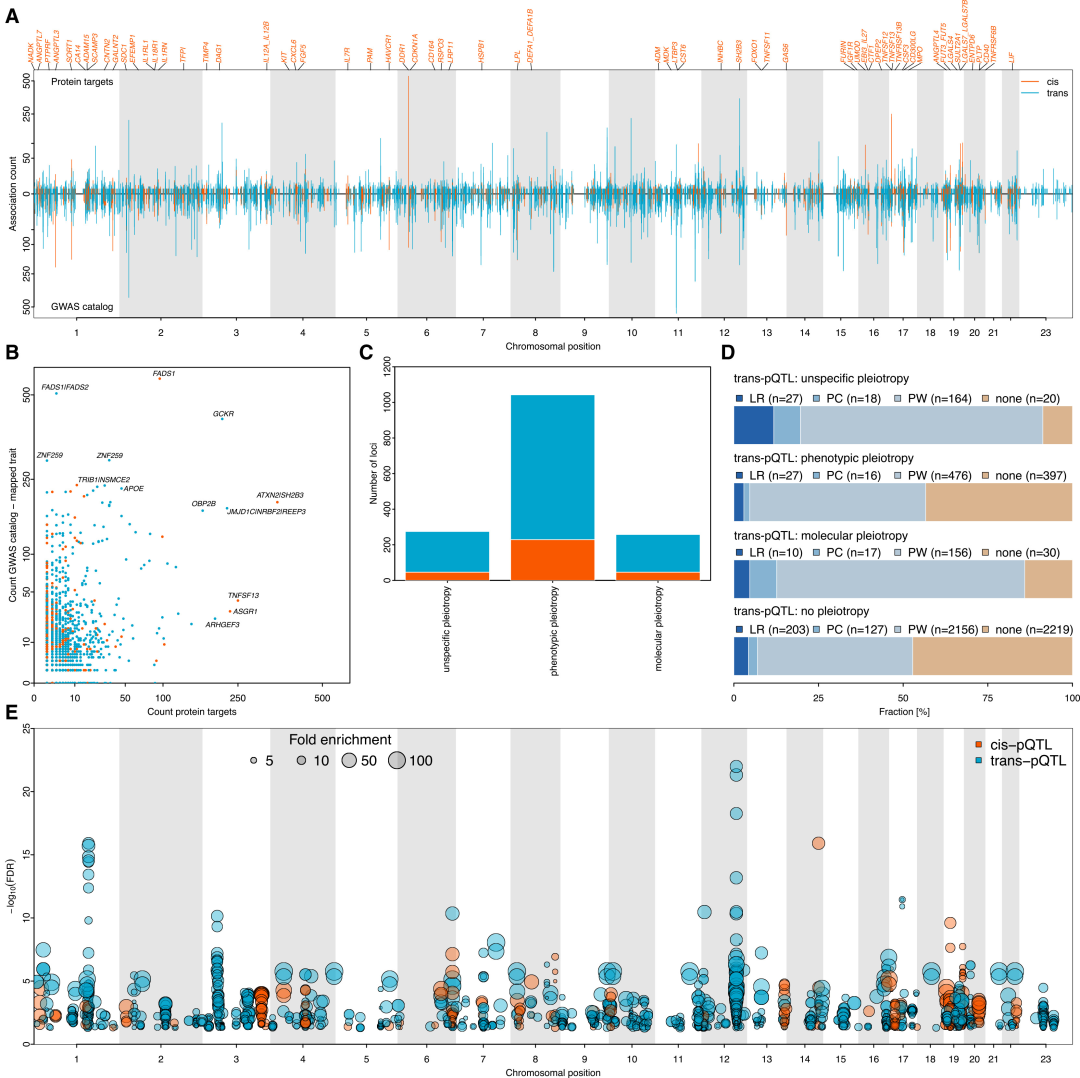
285 pleiotrope pQTLs überlappen sich mit Krankheits-GWAS-Stellen, und ihre assoziierten Proteine sind in spezifischen Signalwegen signifikant angereichert, was neue Hinweise zur Aufklärung der Mechanismen von Krankheits-GWAS-Stellen liefert.
Unterschiede in den Krankheitsphänotypen unter cis- und trans-Regulation
Forscher kombinierten 300 identifizierte, durch cis-pQTL bedingte Protein-Krankheits-Assoziationen mit über 700 Krankheitsdatensätzen aus dem FinnGen-Projekt. Nur 73 dieser Assoziationen lieferten statistische Kolokalisationsanalysen, die sowohl Mendelsche Randomisierung (MR) als auch genetische Risikosignale erfassten.Dies legt nahe, dass ergänzende Beweise erforderlich sind, wenn potenzielle krankheitsverursachende Gene priorisiert werden.
Von den 115 auswertbaren Assoziationen zeigten 31 konsistente cis- und trans-regulatorische Effekte, 41 hatten keine unterstützenden Beweise und 14 zeigten gegensätzliche Effekte, was auf einen signifikanten Unterschied in den Auswirkungen der cis-proximalen und der trans-distalen Regulation auf den Krankheitsphänotyp hinweist.
Protein-Krankheits-Assoziationsanalyse in genetischen Schlussfolgerungen und Beobachtungsstudien
Diese Studie integriert Beobachtungsdaten von 52.164 Teilnehmern der UKBB-Studie und genetische Daten von über 1,29 Millionen Personen aus der PanBio-Datenbank und deckt 517 Krankheiten ab. Von den 193 hochsignifikanten genetischen Assoziationen wurden nur 52 durch Beobachtungsstudien konsistent bestätigt; und von den 52.887 signifikanten Assoziationen in Beobachtungsstudien erhielten nur 0,061 TP3T genetische Evidenz. Bemerkenswerterweise ist das Blut-Furin-Protein eines der wenigen Zielmoleküle, das in genetischen und Beobachtungsstudien konsistent mit Hypertonie, Myokardinfarkt und Vorhofflimmern assoziiert ist, was sein Potenzial für die Arzneimittelentwicklung unterstreicht.
trans-pQTL dient als Leitfaden für die Entdeckung von Krankheitsbiomarkern und die Neuausrichtung von Medikamenten
Mehr als 901 TP3T-Krankheitsprotein-Biomarker (280 von 307 Krankheiten) waren signifikant in trans-pQTL-assoziierten Proteinen angereichert, was bestätigt, dass die Trans-Regulation die zentrale genetische Grundlage von Krankheitsprotein-Biomarkern bildet. Die Studie ergab, dass die TYK2-Gen-Missense-Mutation rs34536443 als trans-pQTL mehrere Entzündungsproteine wie BST2 und CXCL9/10/11 reguliert. Erhöhte Spiegel dieser Proteine sind mit einem erhöhten Risiko für rheumatoide Arthritis, Psoriasis und Autoimmunthyreoiditis assoziiert und liefern genetische Hinweise für die Neupositionierung von TYK2-Inhibitoren zur Behandlung von Autoimmunerkrankungen.
Abschluss
Diese Studie, basierend auf der weltweit größten proteogenomischen Analyse mehrerer Kohorten, hat die genetischen Regulationsmuster des zirkulierenden menschlichen Proteoms systematisch aufgeklärt. Sie überwand die Einschränkungen bisheriger Studien, die sich ausschließlich auf die Cis-Regulation konzentrierten, und enthüllte erstmals umfassend die Schlüsselrolle der transgenen Regulation bei der Steuerung der zirkulierenden Proteinmenge in einer großen Stichprobe. Darüber hinaus nutzte sie maschinelles Lernen, um Effektor-Gene präzise zu lokalisieren und zentrale Signalwege wie die N-Glykosylierung und die Thrombozytenbiologie sowie wichtige regulatorische Bereiche wie Leber und Immunzellen zu identifizieren.
Obwohl diese Studie einige Einschränkungen aufweist – beispielsweise, dass die Proteomik-Technologie nur bestimmte Subtypen und posttranslationale Modifikationen zirkulierender Proteine erfasst und die Hauptpopulation europäischer Abstammung ist und auf weitere ethnische Gruppen ausgeweitet werden sollte –, etabliert sie dennoch ein umfassendes Rahmenwerk, das nicht-kodierende genetische Variationen, zirkulierende Proteine und Krankheitsmechanismen miteinander verknüpft. Dies eröffnet nicht nur eine neue Perspektive zur Aufklärung der molekularen Mechanismen komplexer Erkrankungen, sondern verankert auch wichtige Zielstrukturen wie Plasmafurin und TYK2 durch genetische Evidenz. Dadurch liefert sie hochgradig glaubwürdige genetische Belege für die Entwicklung innovativer Medikamente und die Umwidmung bereits zugelassener Arzneimittel und stellt einen entscheidenden Schritt für die Translation der Proteogenomik von der Grundlagenforschung in die klinische Anwendung dar.