Command Palette
Search for a command to run...
Die Aktivität Der Entworfenen Proteinvarianten Erhöhte Sich Um Das 50-fache! Das Zhou Hao-Team Des AIR Der Tsinghua-Universität Schlug AMix-1 Auf Der Grundlage Bayesscher Flussnetzwerke Vor, Um Ein Skalierbares Und Universelles Proteindesign Zu erreichen.

Derzeit steckt die Forschung im Bereich der Protein-Sockelmodelle noch in der „BERT“-Ära fest, die sich nicht vollständig an die biologischen Eigenschaften von Proteinsequenzen anpassen kann. Zuvor haben KI-Modelle wie AlphaFold und ESM die Entwicklung in mehreren Bereichen deutlich vorangetrieben, darunter Strukturvorhersage, Rückfaltung, Vorhersage funktioneller Eigenschaften, Bewertung von Mutationseffekten und Proteindesign.Allerdings mangelt es diesen Modellen noch immer an skalierbaren und systematischen Methoden, die mit den modernsten großen Sprachmodellen (LLMs) vergleichbar wären, und ihre Fähigkeiten können mit zunehmendem Datenvolumen, Modellmaßstab und Rechenressourcen nicht kontinuierlich verbessert werden.
Die mangelnde Universalität solcher Modelle hat im Bereich des Proteindesigns zu schwer lösbaren Herausforderungen geführt: Die Modelle können die konformationelle Heterogenität von Proteinen nicht erfassen und Vorhersagen zum Proteindesign können nicht über den Rahmen der Trainingsdaten hinausgehen. Und die übermäßige Abhängigkeit von der Übertragung von NLP-Methoden hat dazu geführt, dass es an originellen Architekturentwürfen mangelt, die auf Proteineigenschaften abzielen.
In diesem Zusammenhang schlug die Forschungsgruppe von Zhou Hao am Institute of Intelligent Industries (AIR) der Tsinghua-Universität in Zusammenarbeit mit dem Shanghai Artificial Intelligence Laboratory ein systematisch trainiertes Proteinbasismodell AMix-1 auf Basis eines Bayesschen Flussnetzwerks vor, das einen skalierbaren und allgemeinen Weg für das Proteindesign bietet.Dieses Modell übernahm erstmals die systematische Methodik des „Pretraining Scaling Law“, der „Emergent Ability“, des „In-Context Learning“ und der „Test-time Scaling“ und entwickelte auf dieser Grundlage eine kontextbezogene Lernstrategie auf Basis der multiplen Sequenzalignmentierung (MSA), wodurch Konsistenz im allgemeinen Rahmen des Proteindesigns erreicht und gleichzeitig die Skalierbarkeit des Modells sichergestellt wurde.
Die entsprechenden Forschungsergebnisse wurden auf der arXiv-Plattform unter dem Titel „AMix-1: A Pathway to Test-Time Scalable Protein Foundation Model“ veröffentlicht.
Forschungshighlights:
* Für das auf einem Bayesschen Flussnetzwerk basierende Proteinerzeugungsmodell wurde ein vorhersagbares Skalierungsgesetz festgelegt.
* Das AMix-1-Modell entwickelt spontan ein „perzeptuelles Verständnis“ der Proteinstruktur allein durch Trainingsziele auf Sequenzebene, ohne dass eine explizite strukturelle Überwachung erforderlich ist.
* Das auf der multiplen Sequenzalignmentierung (MSA) basierende kontextuelle Lernframework löst das Alignmentproblem bei der Funktionsoptimierung, verbessert die Argumentations- und Designfähigkeiten des Modells in einem evolutionären Kontext und ermöglicht AMix-1, neue Proteine mit konservierter Struktur und Funktion zu erzeugen;
* Schlagen Sie einen Algorithmus zur Verlängerung der Testzeit anhand der Verifizierungskosten vor, um bei steigenden Verifizierungsbudgets einen neuen evolutionsbasierten Designansatz zu ermöglichen.
Papieradresse:
Folgen Sie dem offiziellen Konto und antworten Sie mit „AMix“, um das vollständige PDF zu erhalten
Weitere Artikel zu den Grenzen der KI:
UniRef50-Datensatz: Vorverarbeitung und iteratives Clustering
Die Forscher verwendeten den vorverarbeiteten UniRef50-Datensatz während des Modell-Vortrainings. Dieser von EvoDiff bereitgestellte Datensatz wurde aus UniProtKB abgeleitet und durch iteratives Clustering (UniProtKB+UniParc → UniRef100 → UniRef90 → UniRef50) aus UniParc-Sequenzen gefiltert., mit 41.546.293 Trainingssequenzen und 82.929 Validierungssequenzen. Sequenzen mit mehr als 1.024 Resten wurden mithilfe einer zufälligen Beschneidungsstrategie auf 1.024 Reste gekürzt, um den Rechenaufwand zu reduzieren und vielfältige Teilsequenzen zu erzeugen. Dieser iterative Prozess gewährleistet eine qualitativ hochwertige, redundante und vielfältige Darstellung der UniRef50-Sequenzen und bietet eine umfassende Abdeckung des Proteinsequenzraums für Proteinsprachenmodelle.
Laden Sie den UniRef50-Datensatz herunter:
Technische Lösungen mit System
AMix-1 bietet einen vollständigen Satz systematischer technischer Lösungen zur Implementierung der Testzeitskalierung für Proteinsockelmodelle:
* Skalierungsgesetz vor dem Training:Es ist klar, wie Parameter, Anzahl der Stichproben und Rechenaufwand ausgeglichen werden müssen, um die Fähigkeiten des Modells zu maximieren.
* Auftauchende Fähigkeit:Es zeigt, dass das Modell im Laufe des Trainings ein „perzeptuelles Verständnis“ der Proteinstruktur entwickelt.
* Kontextbezogenes Lernen:Es löst das Ausrichtungsproblem bei der Funktionsoptimierung und ermöglicht es dem Modell, Argumentation und Design in einem evolutionären Kontext zu erlernen.
* Skalierung der Testzeit:AMix-1 eröffnet einen neuen Ansatz für evolutionsbasiertes Design, da die Verifizierungsbudgets steigen.
Vom Training und der Inferenz bis hin zum Design hat AMix-1 seine Vielseitigkeit und Skalierbarkeit als Protein-Grundmodell unter Beweis gestellt und den Weg für die praktische Umsetzung geebnet.
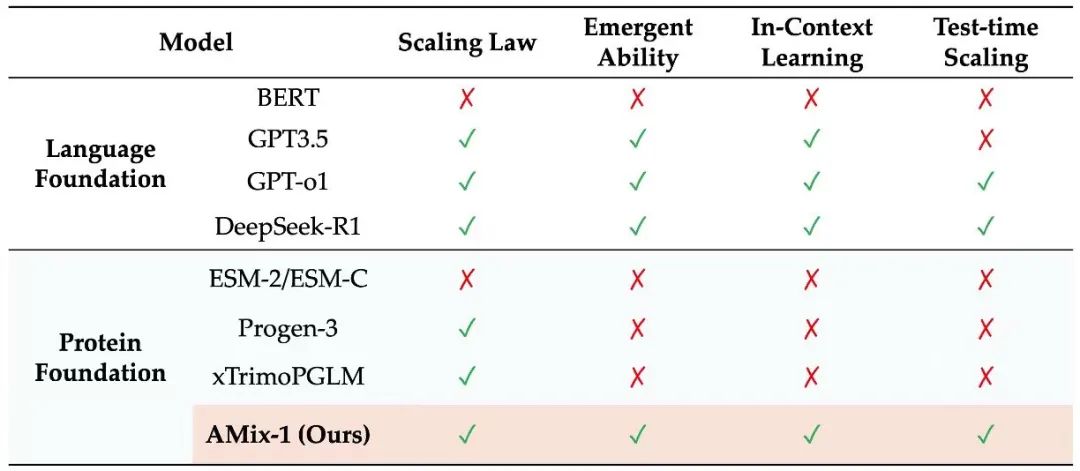
Vortrainings-Skalierungsgesetz: Vorhersagbare Fähigkeiten von Proteinmodellen
Um ein vorhersagbares Skalierungsgesetz für AMix-1 zu erreichen, wurde in dieser Studie eine mehrskalige Modellkombination mit Parametern im Bereich von 8 Millionen bis 1,7 Milliarden im Experiment entworfen und das Training von Gleitkommaoperationen (FLOPs) als einheitlicher Messindikator verwendet, um die Potenzgesetzbeziehung zwischen dem Kreuzentropieverlust des Modells und dem Rechenaufwand genau anzupassen und vorherzusagen.
Den Ergebnissen zufolge ist die Potenzkurve zwischen Modellverlust und Rechenaufwand sehr konsistent, was bestätigt, dass der auf dem Bayes'schen Flussnetzwerk basierende Modelltrainingsprozess sehr vorhersehbar ist.
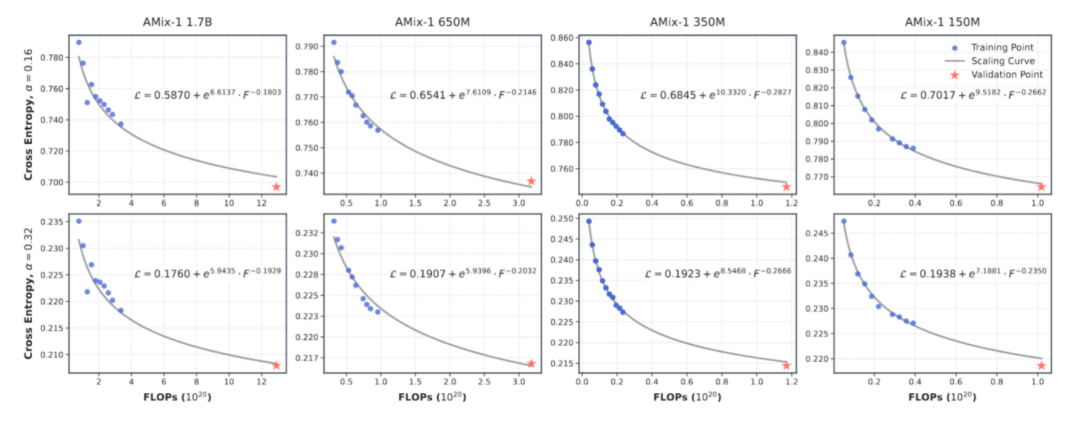
Emergente Fähigkeit: Erreichen erweiterter Modellfähigkeiten
Beim Proteinsequenzlernen basiert die Untersuchung struktureller Emergenz typischerweise auf dem Paradigma „Sequenz-Struktur-Funktion“. Um den Zusammenhang zwischen Optimierungsdynamik und funktionalen Ergebnissen in der Proteinmodellierung zu validieren, analysierte das Forschungsteam emergentes Verhalten aus einer verlustorientierten Perspektive basierend auf einem vorhersagbaren Skalierungsgesetz. Unter Verwendung des prädiktiven Kreuzentropieverlusts als Anker bildeten sie den Trainingsverlust empirisch auf die Proteingenerierungsleistung ab. Die Bewertung der emergenten Fähigkeit des Modells konzentrierte sich in dieser Studie auf drei Aspekte:
* Die Fähigkeit des Modells, Sequenzebenen aus beschädigten Sequenzverteilungen basierend auf Beobachtungen der Sequenzkonsistenz wiederherzustellen;
* Der Übergang von Modellen vom Sequenzverständnis zur strukturellen Machbarkeit aus der Perspektive der Faltbarkeit;
* Beurteilung der Fähigkeit des Modells, strukturelle Eigenschaften auf der Grundlage der strukturellen Konsistenz beizubehalten.
Die relevanten Daten während des AMix-1-Trainings demonstrieren vollständig den Entstehungsprozess der Fähigkeiten des Protein-Basismodells hinsichtlich „Sequenzkonsistenz, Faltbarkeit und Strukturkonsistenz“.Die Daten zeigen, dass alle Fähigkeitsindikatoren des Modells während des Trainings stark mit dem Kreuzentropieverlust korrelieren, was die Möglichkeit bestätigt, die Modellfähigkeit durch Skalierungsgesetz und Kreuzentropieverlust vorherzusagen.Gleichzeitig weist das Modell, wenn es nur mit selbstüberwachten Zielen auf Sequenzebene trainiert wird und keine strukturellen Informationen einführt, immer noch Notfallfähigkeit auf, nachdem der Kreuzentropieverlust auf einen Schwellenwert gesunken ist, und zeigt einen nichtlinearen Übergang zwischen pLDDT und TM-Score.
Kontextbezogenes Lernen: Ein allgemeines Paradigma für das Proteindesign
Durch Computersimulationen überprüften die Forscher den In-Context-Learning-Mechanismus von AMix-1. Die Simulationsexperimente zeigten, dassAMix-1 ist in der Lage, strukturelle oder funktionale Einschränkungen aus Eingabeproben präzise zu extrahieren und zu verallgemeinern, ohne auf explizite Beschriftungen oder strukturelle Überwachung angewiesen zu sein.
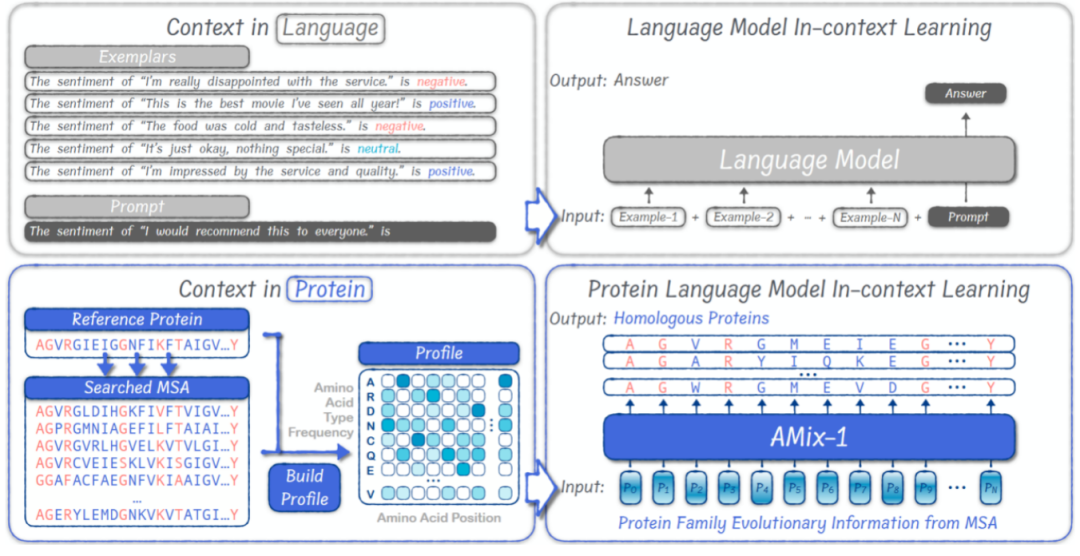
Im Vergleich zum traditionellen Proteindesign, das aufgabenspezifische, individuell angepasste Prozesse erfordert und über kein einheitliches Proteindesign-Framework verfügt, führt AMix-1 einen kontextbasierten Lernmechanismus (ICL) innerhalb eines großen Sprachmodells ein, um struktur- und funktionsorientiertes Proteindesign zu ermöglichen. Experimente haben gezeigt, dass AMix-1 bei Strukturaufgaben neuartige Proteine mit hochgradig konsistenten vorhergesagten Strukturen generieren kann, wobei konventionelle homologe Proteine oder sogar Proteine mit praktisch keiner Homologie als Referenz dienen. Bei Funktionsaufgaben kann AMix-1 hochgradig konsistente Proteasen basierend auf der enzymatischen Funktion und dem durch chemische Reaktionen gesteuerten Design des Eingangsproteins generieren.
Im Rahmen dieses allgemeinen MechanismusDas Modell kann automatisch die gemeinsamen Informationen und Regeln einer bestimmten Proteingruppe ableiten und diese Regeln verwenden, um die Generierung neuer Proteine zu steuern, die den gemeinsamen Regeln entsprechen.Dieser Mechanismus komprimiert eine Gruppe von Protein-MSAs in eine Wahrscheinlichkeitsverteilung auf Positionsebene (Profil), die in das Modell eingegeben wird. Nach einer schnellen Analyse der Struktur und Funktionsregeln der Eingabeproteine kann das Modell neue Proteine generieren, die der Absicht entsprechen.
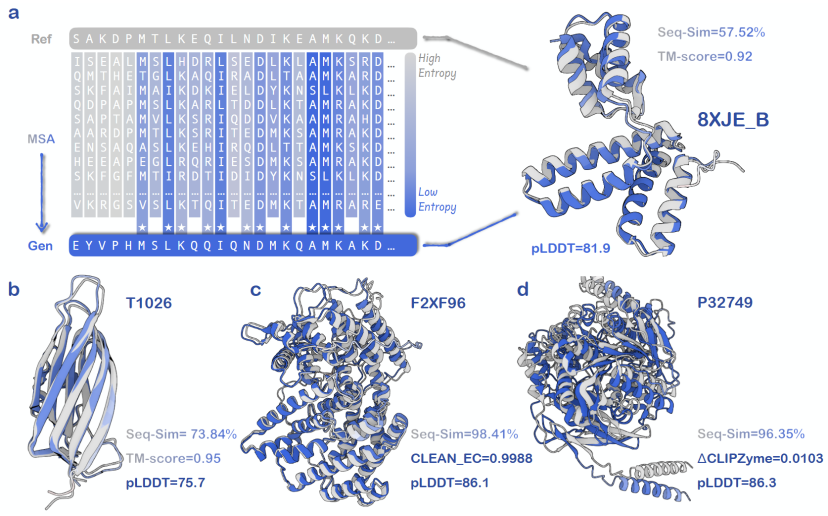
Skalierung der Testzeit: Skalierbare allgemeine Intelligenz
Basierend auf dem Testzeitskalierungsansatz nutzten die Forscher das Proposer-Verifier Framework zum Aufbau von EvoAMix-1. Durch die kontinuierliche Erhöhung des Verifikationsbudgets verbesserten sie die Modellleistung von AMix-1. Neben der Steigerung der Designeffizienz des Modells erreichte das Team auch Skalierbarkeit. Um die Kompatibilität zu gewährleisten, eliminierte das Team außerdem vordefinierte Anforderungen an die Eigenschaften des Verifizierers.
EvoAMix-1 fördert die Erforschung von Proteinsequenzen auf Basis der inhärenten Zufälligkeit probabilistischer Modelle. Durch die Integration aufgabenspezifischer Belohnungsfunktionen aus Computersimulationen oder experimentellem Detektionsfeedback generiert und screent es iterativ Kandidatenproteinsequenzen unter evolutionären Bedingungen. Es ermöglicht eine effiziente, gelenkte Proteinevolution ohne Feinabstimmung des Modells und erzielt so eine robuste und im Testzeitraum skalierbare Leistung im Proteindesign.Bei allen sechs Designaufgaben übertrifft EvoAMix-1 AMix-1 beim In-Context-Learning und verschiedenen starken Basismethoden durchweg.
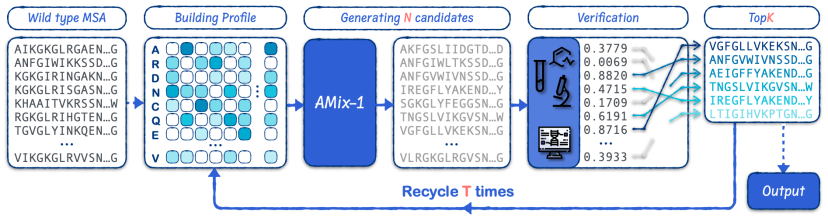
Im Vergleich zur traditionellen Methode zur Generierung neuer Proteinvarianten durch Importance Sampling,EvoAMix-1 aktualisiert keine Modellparameter, sondern erstellt stattdessen eine Vorschlagsverteilung anhand kontextbezogener Beispiele.In jeder Runde verwendet AMix-1 als Hinweise eine Reihe von Mehrfachsequenzalignments (MSAs) oder deren Spektren, die als Eingabebedingungen für ein Proteinbasismodell betrachtet werden, das dann benachbarte Sequenzen abtastet und so effektiv eine neue bedingte Vorschlagsverteilung definiert.
Das Forschungsteam validierte systematisch die Vielseitigkeit und Skalierbarkeit von EvoAMix-1 anhand mehrerer repräsentativer Aufgaben der proteingesteuerten Evolution, darunter die optimale pH- und Temperaturentwicklung von Enzymen, Funktionserhalt und -verbesserung, das Design von Orphan-Proteinen sowie die allgemeine strukturgesteuerte Optimierung. Die experimentellen Ergebnisse belegen die robuste Skalierbarkeit der Testzeitskalierung von EvoAMix-1 und damit die hohe Vielseitigkeit über verschiedene Aufgaben und Ziele hinweg.
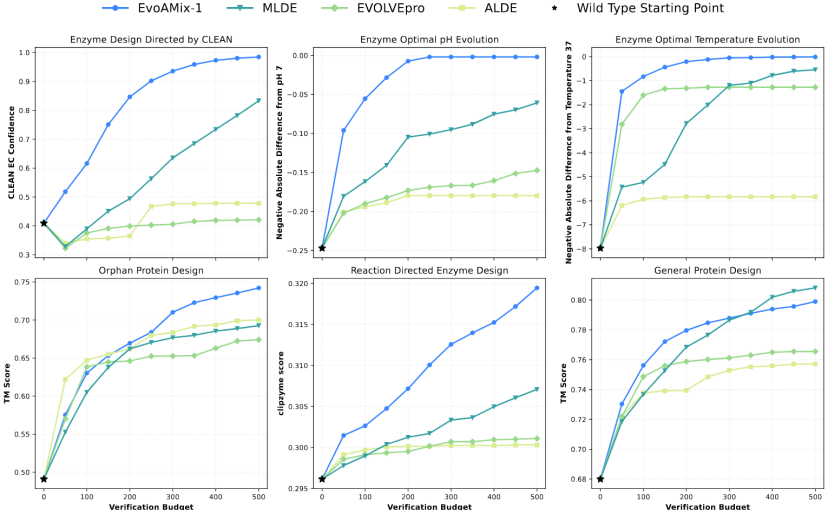
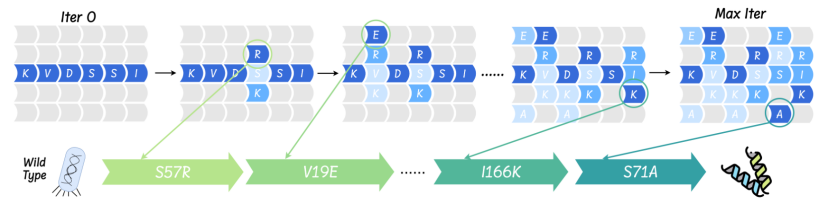
Nassexperiment-Verifizierung: AMix-1 unterstützt die Entwicklung von Protein-AmeR-Varianten mit einer 50-fach erhöhten Aktivität
Die Studie testete die Strategie des „kontextorientierten Designs“ in realen Nassversuchen und bestätigte damit die Vorteile von AMix-1 bei der effizienten Entwicklung hochaktiver AmeR-Varianten. Die Forscher wählten das Zielprotein AmeR aus und verwendeten das AMix-Modell, um 40 Varianten basierend auf der Wahrscheinlichkeitsverteilung der AmeR-Familie zu erzeugen. Die Hemmwirkung jeder Variante wurde durch Experimente mit fluoreszierenden Reportergenen bewertet. Jede Variante enthielt lediglich ≤ 10 Aminosäuremutationen, und je höher der Repressionswert, desto stärker die Funktion. Darüber hinaus schlug die Studie einen Skalierungsalgorithmus für evolutionäre Tests vor, um die Anwendbarkeit von AMix-1 in der gerichteten Proteinevolution zu verbessern, und verifizierte seine Leistungsfähigkeit durch eine Vielzahl von Zielbereichsindikatoren aus Computersimulationen.
Die endgültigen Ergebnisse zeigen, dassDie von AMix-1 generierte optimale Variante weist eine bis zu 50-fache Aktivitätsverbesserung auf und ihre Leistung ist im Vergleich zum aktuellen SOTA-Modell um etwa 77% verbessert.Darüber hinaus ist AMix-1 nicht auf wiederholtes Screening oder manuelles Design angewiesen, sondern wird vollständig automatisch vom Modell generiert.Es wurde ein vollständig geschlossener Kreislauf vom „Modell zum Experiment“ erreicht und der erste Durchbruch bei der Nutzung des funktionalen Proteindesigns durch KI erzielt.
Globale Topologie und Wahrnehmung eröffnen eine neue Dimension im Proteindesign
Derzeit boomt die Forschung zur Integration von KI und Proteindesign. Neben AMix-1 hat auch das geometriebewusste Diffusionsmodell TopoDiff, das von Gong Haipengs Team an der School of Life Sciences der Tsinghua-Universität und Xu Chunfus Team am Beijing Institute of Life Sciences vorgeschlagen wurde, bedeutende Durchbrüche im Proteindesign erzielt.
Herkömmliche Diffusionsmodelle wie RFDiffusion leiden nicht nur unter Abdeckungsverzerrungen bei der Generierung spezifischer Faltungstypen wie Immunglobuline, sondern verfügen auch nicht über quantitative Bewertungsmaße für die globale Topologie des Proteins. Diese Studie, die auf Strukturdatenbanken wie CATH und SCOPe basiert, schlug ein unbeaufsichtigtes System vor, das TopoDiff-Framework. Durch das Erlernen und Ausnutzen globaler, geometrisch bewusster latenter Darstellungen erreicht es eine bedingungslose und kontrollierbare Proteingenerierung auf Basis von Diffusionsmodellen. Diese Studie schlägt ein neues Bewertungsmaß namens „Coverage“ vor, das durch ein zweistufiges Encoder-Diffusionsmodell-Framework die Proteinstruktur in einen globalen geometrischen Bauplan und die Generierung lokaler Atomkoordinaten entkoppelt und so die Forschungsherausforderungen der Proteinfaltungsabdeckung meistert.
Darüber hinaus hat NVIDIA in Zusammenarbeit mit Mila, dem Quebec Institute for Artificial Intelligence in Kanada, die Herausforderung der Langkettenvorhersage mithilfe eines verbesserten All-Atom-Generierungsmodells auf Basis der AlphaFold-Architektur gemeistert. Herkömmliche Methoden haben nicht nur Schwierigkeiten, All-Atom-Strukturen sehr langer Ketten (> 500 Reste) zu generieren, sondern scheitern auch an der Untersuchung nicht-klassischer Faltungskonformationen, wie z. B. membranproteinspezifischer Taschen. Das Forschungsteam führte einen probabilistischen Entscheidungsmechanismus ein, der deterministische Faltungstrajektorien durch Pfadintegral-Sampling aus der Quantenfeldtheorie ersetzt und so die Erfolgsrate des Membranproteindesigns auf 68% erhöht.
Von der geometrischen Erfassung der Proteinfaltung über die Entwicklung langer Ketten mit über 500 Aminosäuren und sprachgesteuertem Proteindesign bis hin zur gezielten Behandlung „nicht medikamentös behandelbarer“ IDPs – KI erweitert die Möglichkeiten des Proteindesigns und schafft ein neues Paradigma für die Forschung auf diesem Gebiet. KI-gesteuertes Proteindesign dürfte künftig noch mehr Möglichkeiten für die Entwicklung innovativer Therapeutika, Enzyme und Biomaterialien eröffnen.
Referenzlinks: