Command Palette
Search for a command to run...
Die Durch 56% Verbesserte Empfindlichkeit, CUHK/Fudan/Yale Und Andere Schlugen Gemeinsam Eine Neue Methode Zur Erkennung Von Proteinhomologen Vor

Eiweiß ist die materielle Grundlage des Lebens und der Hauptträger der Lebensaktivitäten. Im postgenomischen Zeitalter ist mit der Entwicklung der Proteinbestimmungstechnologie die Größe der Proteinsequenzdatenbanken explosionsartig angestiegen. Um ein tieferes Verständnis der Vielfalt und Funktionen von Proteinen zu erlangen, ist in der Biologie die Proteinidentifizierung besonders wichtig.
Im Prozess der Proteinerkennung ist die Identifizierung der Proteinsequenzhomologie eine der wichtigsten Aufgaben.Es kann Wissenschaftlern helfen, die evolutionären Beziehungen, strukturellen Eigenschaften und Funktionen von Proteinen zu verstehen. Obwohl herkömmliche Methoden zur Protein-Sequenzausrichtung in vielen Fällen gute Ergebnisse liefern, sind sie mit entfernten Homologen nicht zurechtgekommen. Diese entfernten Homologe werden bei routinemäßigen Alignments aufgrund ihrer geringen Sequenzähnlichkeit oft übersehen, was das umfassende Verständnis der Forscher hinsichtlich der Vielfalt und Komplexität von Proteinen einschränkt.
Um die Schwachstellen der Forschung zur entfernten Proteinhomologie zu lösen, haben Li Yu von der Chinesischen Universität Hongkong zusammen mit Sun Siqi, einem jungen Forscher vom Labor für intelligente komplexe Systeme der Universität Fudan und dem Shanghai Artificial Intelligence Laboratory, sowie Mark Gerstein von der Yale University ein ultraschnelles und hochempfindliches Framework zur Homologieerkennung vorgeschlagen – den Dense Homology Retriever (DHR), der auf Proteinsprachenmodellen und Dense-Retrieval-Technologie basiert.
DHR kann entfernte Homologe identifizieren, die tief in der Sequenz verborgen sind, ohne sich auf die traditionelle Sequenzausrichtung verlassen zu müssen. Dies geschieht durch die leistungsstarken Funktionen der dualen Encoderstruktur und des Proteinsprachenmodells, was eine beispiellose Geschwindigkeit und Empfindlichkeit bei der Homologidentifizierung ermöglicht. Die Forschungsarbeit wurde in der international renommierten Fachzeitschrift Nature Biotechnology unter dem Titel „Fast, sensitive detection of protein homologs using deep dense retrieval“ veröffentlicht.
Forschungshighlights:
* Im Vergleich zu früheren Methoden verbessert DHR die Empfindlichkeit um mehr als 10% und verbessert die Empfindlichkeit um mehr als 56% auf Superfamilienebene für Proben, die mit auf Ausrichtung basierenden Methoden schwer zu identifizieren sind
* Der DHR-Code fragt Sequenzen und Datenbanken 22-mal schneller ab als herkömmliche Methoden wie PSI-BLAST und DIAMOND und 28.700-mal schneller als HMMER

Papieradresse:
https://doi.org/10.1038/s41587-024-02353-6
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Erstellen von Datensätzen in mehreren Dimensionen, um ein breiteres Spektrum an Proteinsequenzen zu untersuchen
Der in dieser Studie erstellte Trainingssatz umfasst 2 Millionen sorgfältig aus UR90 ausgewählte Abfragesequenzen.Mithilfe des JackHMMER-Algorithmus suchte diese Studie iterativ nach Kandidatensequenzen in Uni-Cluster30 und richtete die Kandidatensequenzen mit der multiplen Sequenzalignmentierung (MSA) aus. Jedes MSA enthielt 1.000 Homologe, wodurch sichergestellt wurde, dass nur die am stärksten verwandten Sequenzen beibehalten wurden. Nach einer strengen Prüfung wurde JackHMMER erneut eingesetzt, um die verschiedenen erhaltenen Sequenzen zu verarbeiten, und verwendete dieselben Hyperparametereinstellungen wie AF2 (AlphaFold 2), um einen fairen Vergleich zu ermöglichen.
Bei der Untersuchung großer Datensätze wurde der Datensatz BFD/MGnify ausgewählt.Dabei handelt es sich um eine riesige Datenbank mit etwa 300 Millionen Proteinen, die die Erforschung eines größeren Spektrums an Proteinsequenzen ermöglicht.
Die DHR-Methode: eine ultraschnelle und empfindliche Proteinhomologie-Suchpipeline
Die Kernidee der DHR-Methode besteht darin, Proteinsequenzen in dichte Einbettungsvektoren zu kodieren, um so die Ähnlichkeit zwischen Sequenzen effektiv berechnen zu können.Insbesondere wurde in dieser Studie der Sequenzencoder durch Initialisierung von ESM und Integration kontrastiver Lerntechniken effektiv trainiert, wodurch die Voraussetzungen für die Konstruktion eines Proteinsprachenmodells geschaffen wurden und DHR effektiver zum Abrufen von Homologen verwendet werden konnte.
Wie in Abbildung a unten gezeigt, ist die Studie mit Abschluss der Dual-Encoder-Trainingsphase in der Lage, qualitativ hochwertige Offline-Protein-Sequenzeinbettungen zu generieren. Die Studie verwendete dann diese Einbettungen und Ähnlichkeitssuchalgorithmen, um Homologe für jedes Abfrageprotein abzurufen. Durch die Angabe der Ähnlichkeit als Abrufmetrik können ähnliche Proteine genauer gefunden werden als mit herkömmlichen Methoden, und die Ähnlichkeit zwischen zwei Proteinen kann für weitere Analysen verwendet werden. Schließlich erstellte JackHMMER die MSA der abgerufenen Homologe und die Studie erhielt die DHR-Technologie, mit der Homologe schnell und effektiv entdeckt werden können.
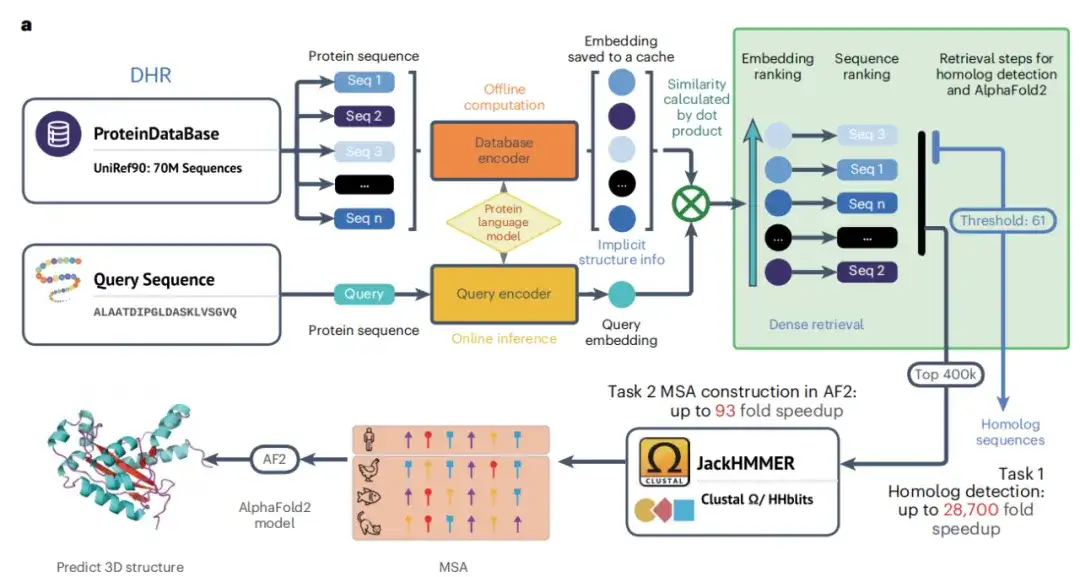
Darüber hinaus wurde im Rahmen der Studie auch ein Hybridmodell DHR-Meta entwickelt, das durch die Kombination von DHR und AF2-Standard die einzelnen Pipelines auf CASP13DM- (Domänensequenz) und CASP14DM-Zielen übertraf.
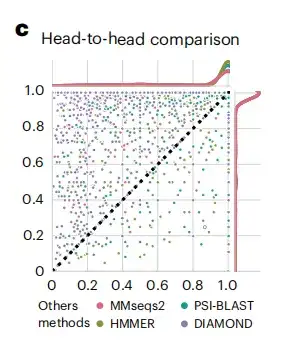
Nach Erhalt der generierten Proteineinbettungen bewertete die Studie die Leistung von DHR durch Vergleich mit Methoden des Standard-SCOPe-Datensatzes (Structure Classification of Proteins).Wie in Abbildung c unten gezeigt, ist die Empfindlichkeit der DHR-Daten besser als bei anderen Methoden.
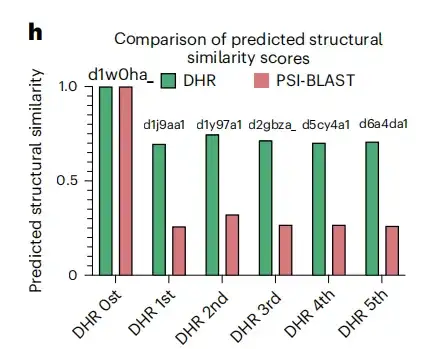
Darüber hinaus lieferten, wie in Abbildung h unten gezeigt, im spezifischen Beispiel der d1w0ha-Abfrage weder PSI-BLAST noch MMseqs2 übereinstimmende Ergebnisse, aber DHR rief 5 Homologe ab, die in SCOPe ebenfalls in dieselbe Familie wie d1w0ha eingeordnet wurden. Dies bedeutet, dass DHR mehr Strukturinformationen erfassen kann. Im Vergleich zu herkömmlichen Methoden wie PSI-BLAST, MMseqs2, DIAMOND und HMMER hat DHR die meisten Homologe erkannt (Sensitivität von 93 %).Dies zeigt, dass DHR in der Lage ist, umfangreiche Strukturinformationen zu integrieren und in vielen Fällen eine Sensitivität von 100 % zu erreichen.
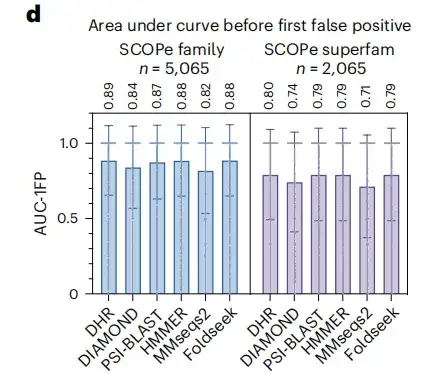
Um die Glaubwürdigkeit der Ergebnisse zu stärken, wurde in diese Studie auch ein weiterer Standardindikator einbezogen: der Bereich unter der Kurve vor dem ersten FP. Die Ergebnisse zeigen, dass DHR, wie in Abbildung d unten dargestellt, einen Wert von 89% erreicht.Mittlerweile zeigten auch andere Methoden eine mit DHR vergleichbare Leistung, ihre Ausführungszeit war jedoch deutlich länger.Als wir uns weiter auf die Ebene der Superfamilie bewegten, um den anspruchsvolleren Satz entfernter Homologe zu analysieren, kam es bei allen Methoden zu einem erheblichen Leistungsabfall, mit einem Gesamtabfall von ungefähr 10%. Trotzdem behält DHR seine führende Leistung mit einem AUC-1FP-Wert von bis zu 80% bei.
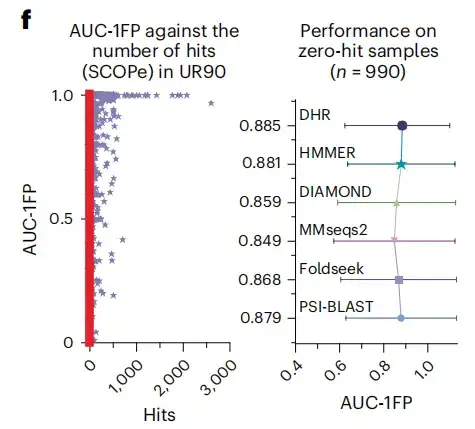
Die Studie ergab außerdem, dass beim Vergleich der SCOPe-Datenbank und von UniRef90 mit BLAST die meisten Proben weniger als 100 Übereinstimmungen ergaben und sogar etwa 500 Proben keine Übereinstimmungen ergaben, was darauf hindeutet, dass es sich bei diesen Proben um „ungesehene“ Strukturen im Trainingsdatensatz handelte. Im Gegensatz dazu erzielt DHR für diese Strukturen immer noch qualitativ hochwertige Vorhersagen und erreicht einen AUC-1FP-Score von 89%.Dies zeigt die Fähigkeit des DHR, völlig neue Daten zu verarbeiten.
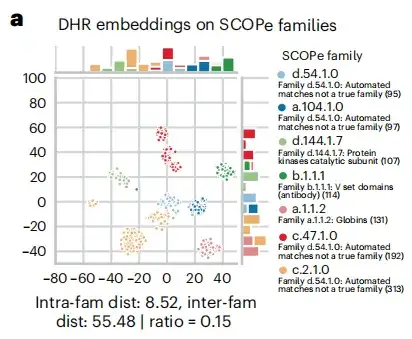
Während des Homologiesuchprozesses stellte die Studie fest, dass die DHR-Sequenzeinbettung, wie in Abbildung a unten dargestellt, eine große Menge an Strukturinformationen enthält und dass die Genauigkeit von DHR beim Abrufen homologer Substanzen sogar die von strukturbasierten Ausrichtungsmethoden übertrifft. Basierend auf diesem Ergebnis,Diese Studie enthüllte außerdem die Korrelation zwischen der Sequenzähnlichkeitsrangfolge und der strukturellen Ähnlichkeit von DHR.
Forschungsergebnisse: DHR ist präziser und effektiver und kann hochwertige MSA auf großen Datensätzen erstellen
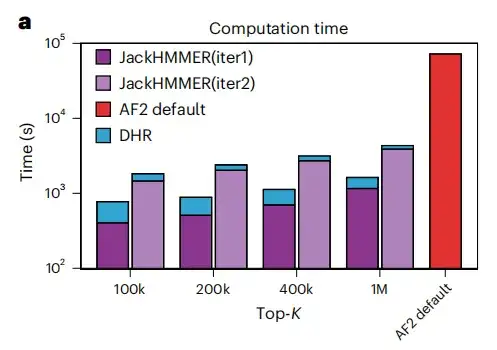
Wir haben MSAs aus JackHMMER mithilfe von Homologen erstellt, die von DHR bereitgestellt wurden, und sie mit der AF2-Standardpipeline verglichen. Wie in Abbildung a unten gezeigt, ist die durchschnittliche Laufgeschwindigkeit aller Konfigurationen von DHR + JackHMMER schneller als die des gewöhnlichen JackHMMER von AF2. Darüber hinaus überlappt sich DHR beim Erstellen von MSA auf UniRef90 um etwa 80% mit JackHMMER.Dies lässt darauf schließen, dass viele nachgelagerte Aufgaben im Zusammenhang mit MSA mithilfe von DHR ausgeführt werden können und zu ähnlichen, aber schnelleren Ergebnissen führen.
Wie in den Abbildungen e und f unten gezeigt, besteht ein weiterer Vorteil von DHR darin, dass es in einer konstanten Zeit die gleiche Anzahl von Homologen unterschiedlicher Länge konstruieren kann, während JackHMMER linear skaliert. Darüber hinaus kann DHR im Vergleich zu AF2 mehr Homologe und MSA für die Abfrageeinbettung bereitstellen. Diese Ergebnisse zeigen, dassDHR ist ein vielversprechender Ansatz für alle Kategorien des MSA-Baus.
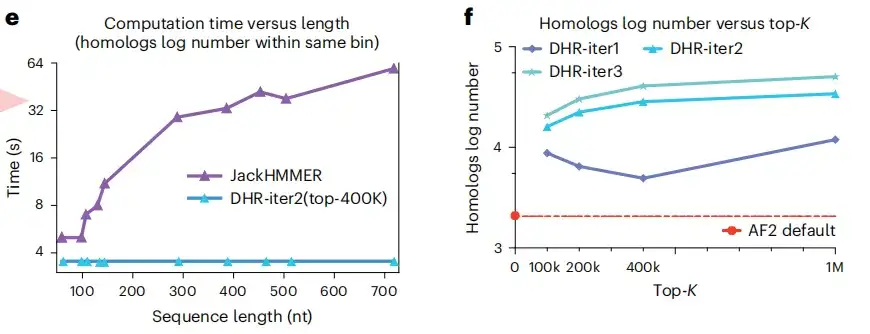
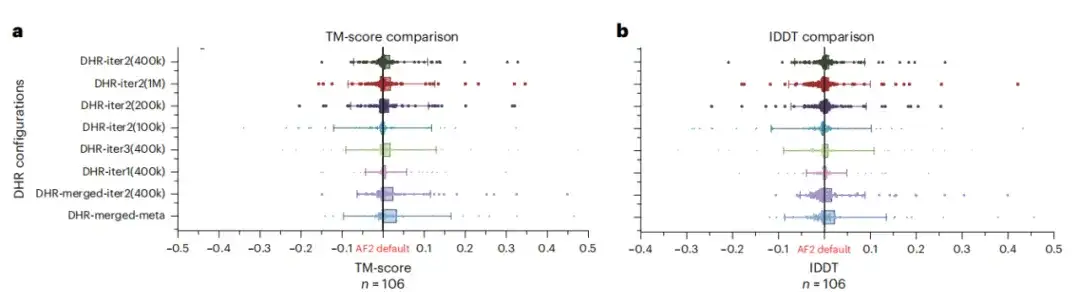
Obwohl DHR in der Lage ist, verschiedene MSAs zu erstellen, wurde in dieser Studie weiter analysiert, ob es als Ergänzung zum AF2-Basis-MSA dienen könnte. Die Forschungsergebnisse zeigen, dass, wie in den Abbildungen a und b unten dargestellt, die Leistung der Kombination aller MSA und AF2 unter verschiedenen DHR-Einstellungen am besten ist.Dies bedeutet, dass DHR die MSA-Pipeline von AF2 schnell und präzise auffüllen kann.
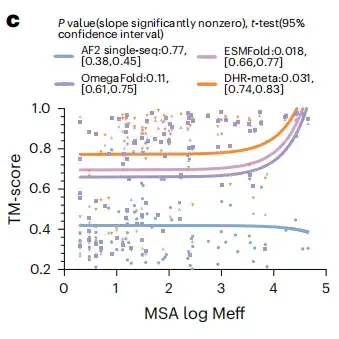
Um die potenziellen Vorteile großer Sprachmodelle für die Vorhersage von Proteinstrukturen zu untersuchen, wurde in dieser Studie untersucht, ob der Ersatz von MSA durch ein großes Sprachmodell auf allen CASP14DM-Zielen zu besseren Ergebnissen führen würde. Wie in Abbildung c unten gezeigt, kann das Sprachmodell im einfachen Fall mit einer großen Anzahl verfügbarer MSAs genauso viele Informationen übermitteln wie die MSAs. Mit zunehmender Sequenzlänge wird die Leistung von DHR-Meta jedoch immer besser und übertrifft ESMFold in fast allen Fällen. Dies bedeutet, dass im Vergleich zu sprachmodellbasierten MethodenDas MSA-basierte Modell kann die Genauigkeit und Effektivität von Vorhersagen erheblich verbessern.
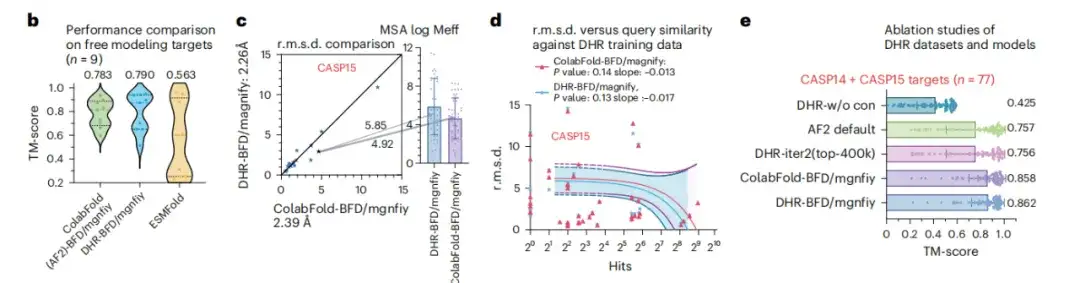
Um die Skalierbarkeit von DHR in großen Datensätzen zu untersuchen, wurde in dieser Studie eine eingehende Analyse von DHR basierend auf BFM/MGnify durchgeführt. Wie in Abbildung b unten gezeigt, kann sich DHR im komplexen Szenario der Vorhersage der Struktur von FM-Zielen durch die Generierung aussagekräftigerer MSAs hervortun, wobei die ColabFold-Methode unter Verwendung von MMseqs2 zur Konstruktion von MSAs eine um 0,007 TM-Scores bessere Leistung erzielt.
In Abbildung 2c zeigt DHR eine leichte Leistungsverbesserung im Vergleich zu ColabFold-MMseqs2. Abbildung d unten zeigt auch, dass nach dem Ähnlichkeitstest von CASP14 und SCOPe festgestellt wurde, dass DHR sich nicht einfach an die Ergebnisse der Abfrage oder Treffer erinnerte, sondern eine umfassende Ähnlichkeitsbewertung aller Ziele durchführte. Diese Ergebnisse beweisen, dassDHR ermöglicht die Konstruktion von MSAs ungeordneter Proteine auf umfangreichen Suchdatensätzen mit hoher Diversität.
Junge Kräfte auf dem Gebiet der Proteinstrukturvorhersage
Es besteht kein Zweifel, dass die Vorhersage von Proteinstrukturen bei Anwendungen wie der Arzneimittelentwicklung und dem Antikörperdesign eine wichtige Rolle spielt. KI könnte der Schlüssel zur Lösung des historischen Problems der begrenzten Genauigkeit bei der Vorhersage von Proteinstrukturen sein. In diesem wichtigen Bereich haben sich unter einheimischen wissenschaftlichen Forschungsteams nach und nach hundert Denkschulen herausgebildet und aufstrebende junge Forscher sind zu einer Kraft geworden, die man nicht ignorieren kann. Li Yu und Sun Siqi, die die oben genannten Forschungsergebnisse leiteten, gehören beide zu den Besten.

Li Yu erhielt 2015 seinen Bachelor-Abschluss (mit Auszeichnung) in Biowissenschaften von der Bei Shizhang Elite Class der University of Science and Technology of China, im Dezember 2016 seinen Master-Abschluss in Informatik von der King Abdullah University of Science and Technology (KAUST) in Saudi-Arabien und 2020 seinen Doktortitel in Informatik von derselben Universität.
Im Dezember desselben Jahres kehrte er nach China zurück und wurde Assistenzprofessor an der Fakultät für Informatik und Ingenieurwesen der Chinesischen Universität Hongkong, wo er die Gruppe „Künstliche Intelligenz im Gesundheitswesen“ (AIH) leitete. Er führte eingehende Forschungen an der Schnittstelle zwischen maschinellem Lernen, Gesundheitswesen und Bioinformatik durch und leitete das Team bei der Entwicklung neuer Methoden des maschinellen Lernens zur Lösung rechnergestützter Probleme in der Biologie und im Gesundheitswesen, insbesondere strukturierter Lernprobleme.
Zu den Bereichen Biologie und Gesundheitswesen, in denen er sich intensiv engagiert, sagte Li Yu: „Mein langfristiges Ziel ist es, das Gesundheitssystem zu verbessern und der Gesellschaft durch die Verbesserung der Gesundheit und des Wohlbefindens der Menschen einen direkten Nutzen zu bringen.“Erwähnenswert ist, dass er auch in die Forbes Asia-Liste „30 unter 30“ (Gesundheitswesen und Wissenschaft) 2022 aufgenommen wurde.

Sun Siqi hat im globalen Wettbewerb zur Vorhersage von Proteinstrukturen hervorragende Ergebnisse erzielt und ist derzeit Nachwuchsforscher am Labor für Grundlagentheorie und Schlüsseltechnologien für intelligente komplexe Systeme und am Shanghai Artificial Intelligence Laboratory der Universität Fudan.Er engagiert sich in der Anwendungsforschung des Deep Learning in interdisziplinären Bereichen wie den Biowissenschaften und der Verarbeitung natürlicher Sprache und konzentriert sich auf die Verbesserung der Genauigkeit und Geschwindigkeit von Modellen sowie die Lösung spezifischer Probleme bei der Implementierung von Modellen.
Im Bereich der Proteinvorhersage konzentriert er sich auf die Vorhersage der Struktur und Sequenz von Proteinen durch Deep-Learning-Modelle und auf das Trainieren von Modellen zur Erkennung von Mustern und Regelmäßigkeiten in Sequenzen, um die Sequenz und Faltung von Proteinen vorherzusagen. Dadurch verbessert er die Genauigkeit und Effizienz der Protein-De-novo-Sequenzierung und Strukturvorhersage und schafft neue Möglichkeiten für die Arzneimittelentwicklung und Krankheitsbehandlung.
Im heimischen AI4S-Bereich sind immer mehr junge Kräfte aktiv. Es ist abzusehen, dass die KI-Technologie im Bereich der Proteinstrukturvorhersage eine wichtigere Rolle spielen wird, doch der Weg dorthin ist lang und beschwerlich. Es ist erfreulich, dass das inländische wissenschaftliche Forschungsteam einen beharrlichen Forschergeist und Innovationsfähigkeit bewiesen hat. Sie haben nicht nur intensiv an der Algorithmusoptimierung und dem Modellaufbau gearbeitet, sondern auch eingehende Untersuchungen in den Bereichen Datenverarbeitung, experimentelle Verifizierung usw. durchgeführt, um die Wissenschaftlichkeit und Praktikabilität der Forschungsergebnisse sicherzustellen. Diese Bemühungen werden nach und nach in praktische Anwendungen umgesetzt und verleihen Bereichen wie der medizinischen Forschung und Entwicklung sowie der Biotechnologie neue Vitalität und Hoffnung.
Abschließend empfehle ich eine akademische Austauschaktivität!
Zur dritten Live-Übertragung von Meet AI4S war Zhou Ziyi eingeladen, ein Postdoktorand am Institut für Naturwissenschaften der Shanghai Jiao Tong University und dem Shanghai National Center for Applied Mathematics. Klicken Sie hier, um einen Termin für die Live-Übertragung zu vereinbaren!
https://hdxu.cn/6Bjomhdxu.cn/6Bjom