Command Palette
Search for a command to run...
اقترحت جامعة تورنتو وآخرون dnaHNet، الذي يحسن سرعة الاستدلال بمقدار 3 مرات ويقلل التكلفة الحسابية لتعلم الجينوم بما يقرب من 4 مرات.

الجينوم هو حامل جميع المعلومات الوراثية للكائن الحي، ويحدد وظيفة الخلية، والتطور الفردي، واتجاه تطور الأنواع.يشكل "تركيب الحمض النووي" المخفي داخل التسلسل القواعد الأساسية التي تحكم الحياة، وهو أحد المشكلات الأساسية التي يحتاج علم الأحياء الحديث إلى حلها بشكل عاجل.إن فهم هذه القواعد النحوية لا يرتبط فقط بالمعرفة العلمية الأساسية، بل يؤثر أيضًا بشكل مباشر على تطوير التطبيقات الرئيسية مثل تشخيص الأمراض وتطوير الأدوية وعلم الأحياء التركيبي.
في السنوات الأخيرة، أصبحت النماذج الأساسية المدربة مسبقًا على بيانات تسلسلية واسعة النطاق مسارًا مهمًا لحل هذه المشكلة. ومع التحسن المستمر في القدرة الحاسوبية وحجم البيانات ومعايير النموذج، أظهرت هذه النماذج اتجاهًا تصاعديًا في الأداء مشابهًا لقانون التوسع. وقد توسعت معايير نماذج مثل Nucleotide Transformer وEvo لتصل إلى مليارات المعايير، وتم تدريبها على تسلسلات من مختلف الأنواع، محققةً تقدمًا ملحوظًا في مهام مثل التنبؤ بتأثير التباين وتحليل العناصر التنظيمية.
مع ذلك، فإن تسلسلات الحمض النووي هي في الأساس سلاسل متصلة من النيوكليوتيدات ذات حدود غير واضحة، وهو اختلاف جوهري عن اللغة الطبيعية. النموذجان الرئيسيان المستخدمان حاليًا في النمذجة هما:يمثل كل من تجزئة الكلمات الثابتة ونمذجة مستوى النوكليوتيدات المفردة مقايضة واضحة بين القدرة التعبيرية والكفاءة الحسابية:قد يُلحق الأول ضرراً بالوحدات الوظيفية البيولوجية، بينما يُكبّد الثاني تكاليف حسابية باهظة. لذا، أصبح تحقيق توازن أفضل بين الجدوى الحسابية والدقة البيولوجية تحدياً رئيسياً. ولا يزال تجزئة الكلمات الديناميكية، كحل محتمل، بحاجة إلى دراسة منهجية.
وفي هذا السياق،نموذج dnaHNet، الذي اقترحته بشكل مشترك جامعة تورنتو، ومعهد فيكتور للذكاء الاصطناعي في كندا، ومعهد آرك في الولايات المتحدة، من بين مؤسسات أخرى،يُقدّم هذا نهجًا جديدًا لتجاوز المعوقات المذكورة آنفًا. وقد نُشرت نتائج البحث ذات الصلة، بعنوان "dnaHNet: نموذج أساسي قابل للتطوير وهرمي لتعلم تسلسل الجينوم"، كنسخة أولية على موقع arXiv.
أبرز ما جاء في البحث
* تتجاوز الكفاءة الحسابية لـ dnaHNet كفاءة StripedHyena2، وسرعة الاستدلال الخاصة بها أسرع بأكثر من 3 مرات من Transformer.
* نقترح استراتيجيات تدريب مثل جدولة نسبة الضغط وموازنة المشفر والمفكك.
* وصل إلى الطليعة في مهام العينات الصفرية مثل التنبؤ بتأثير التباين وتصنيف ضرورة الجينات.
* يمكنها تعلم تجزئة الكلمات البيولوجية المعتمدة على السياق والتكيف مع المناطق الوظيفية مثل الكودونات والمحفزات والمناطق بين الجينات.

عنوان الورقة:
https://arxiv.org/abs/2602.10603
تابع حسابنا الرسمي على WeChat وأجب بكلمة "dnaHNet" في الخلفية للحصول على ملف PDF كامل.
تصميم مجموعات بيانات جينومية متعددة المستويات لتدريب النماذج وتقييمها
لدعم تدريب النموذج وتقييم النظام، قامت هذه الدراسة بإنشاء نظام بيانات متعدد الطبقات.تأتي بيانات التدريب المسبق من مجموعة فرعية معالجة من قاعدة بيانات تصنيف الجينوم (GTDB).اتبعت العملية بدقة إجراءات الترشيح ومراقبة الجودة وإزالة التكرار لمجموعة بيانات OpenGenome الخاصة بنموذج Evo. وشملت معايير الفرز مؤشرات رئيسية مثل سلامة التجميع ومستوى التلوث ومحتوى الجينات الدالة؛ وبعد الفرز، تم الاحتفاظ بجينوم واحد فقط يمثل كل مجموعة على مستوى الأنواع.
تغطي مجموعة البيانات النهائية 85205 من بدائيات النوى وتحتوي على 17648721 تسلسلًا.يبلغ العدد الإجمالي للنيوكليوتيدات حوالي 144 مليار. تم استخراج جميع التسلسلات من الجينوم الكامل وتقسيمها إلى أجزاء غير متداخلة يصل طولها إلى 8192 نيوكليوتيدًا.
من حيث التقييم، قام الباحثون بإنشاء مجموعة اختبار من ثلاثة أبعاد متكاملة لفحص قدرات النموذج بشكل شامل. أولاً،على مستوى لياقة الترميز المحليتم استخدام اثنتي عشرة مجموعة بيانات تجريبية على مستوى النيوكليوتيدات، بإجمالي 21250 نقطة بيانات، مستمدة من E. coli K12 في MaveDB لتقييم قدرة النموذج على توصيف قواعد الترميز المحلية ومشهد لياقة البروتين.
ثانيًا،فيما يتعلق بالتقييم الوظيفي على نطاق الجينوم الكاملاستنادًا إلى قاعدة بيانات الجينات الأساسية (DEG)، تم إنشاء علامات أساسية ثنائية لـ 62 نوعًا من البكتيريا. تم الحصول على التسلسلات والتعليقات ذات الصلة من المركز الوطني لمعلومات التقانة الحيوية (NCBI)، واعتُبرت مطابقة التسلسل التي تزيد عن 99% مع اسم مدخل DEG معيارًا لتصنيف الجينات الأساسية. نتج عن ذلك 185,226 نقطة بيانات، استُخدمت لتقييم قدرة النموذج على دمج التبعيات بعيدة المدى والسياق الجينومي.
أخيرا،من حيث قابلية التفسير الهيكلي،باستخدام جينوم بكتيريا Bacillus subtilis كمثال، تم تقسيم التسلسل إلى مناطق وظيفية مختلفة من خلال دمج بياناته الوظيفية. وتم التحقق من قدرة النمذجة الهيكلية من خلال تحليل مدى تطابق نتائج تجزئة النموذج مع البنى البيولوجية الحقيقية.
نموذج dnaHNet: نموذج حدودي انحداري ذاتي بدون تجزئة الكلمات
dnaHNet هو نموذج قائم على الجينوم لا يتطلب مجزئًا صريحًا.يكمن المفتاح في آلية "التجزئة الديناميكية"، التي تسمح للنموذج بتعلم الوحدات الهيكلية في التسلسل من تلقاء نفسه.يتجنب هذا التصميم تجزئة الأجزاء الوظيفية البيولوجية عن طريق تجزئة الكلمات الثابتة ويخفف من العبء الحسابي لنمذجة النيوكليوتيدات، وبالتالي تحقيق توازن أفضل بين القدرة التعبيرية والكفاءة الحسابية.
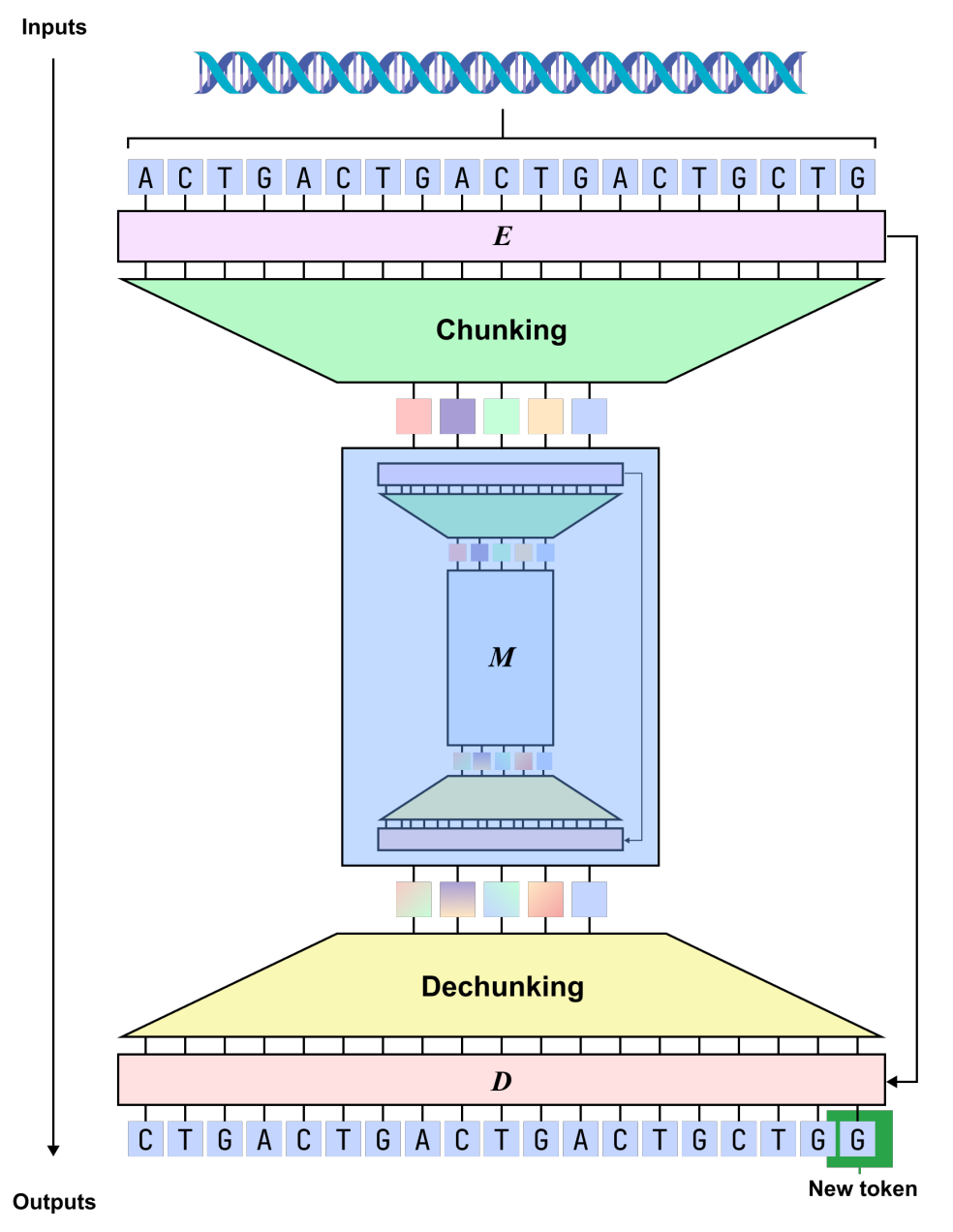
فيما يتعلق بنمذجة الشكل،يوحد dnaHNet عملية تعلم الجينوم في مهمة تنبؤ تسلسلية ذاتية التراجع، والتي تتنبأ بالنيوكليوتيد التالي بناءً على السياق الحالي.تعتمد البنية العامة هيكلاً هرمياً، حيث تتكون كل طبقة من مشفر وشبكة أساسية ومفكك شفرة: يحدد المشفر المواقع في التسلسل حيث تتغير المعلومات بشكل كبير (مثل حدود الكودون أو المناطق التنظيمية) من خلال آلية توجيه، ويضغط التسلسل إلى تمثيل كتلة ضمني وفقًا لذلك؛ تعتمد الشبكة الأساسية بنية هجينة تجمع بين Mamba و Transformer لنمذجة التسلسل المضغوط، مع مراعاة كل من التبعيات بعيدة المدى وتفاعلات المعلومات الرئيسية؛ ثم يقوم المفكك بإعادة أخذ عينات التمثيل إلى دقة النيوكليوتيدات ويخرج نتائج التنبؤ.
انطلاقاً من هذا الأساس، خضعت شبكة dnaHNet لعدة تحسينات رئيسية لبيانات الجينوم. أولاً، فيما يتعلق بتخصيص المعلمات، تم تخصيص ما يقرب من 301 TP3T من سعة النموذج للمشفّر والمفكك لتعزيز القدرة على توصيف البنى المحلية.
ثانيًا،يتضمن تصميم ضغط ثنائي المراحل متعدد الطبقات:تركز المرحلة الأولى على استخلاص الأنماط قصيرة النطاق (مثل الكودونات)، بينما تُصمّم المرحلة الثانية نطاقًا أوسع من البنى الوظيفية، ما يحقق توازنًا بين كفاءة الضغط ودقة المعلومات. علاوة على ذلك، تتضمن عملية التدريب دالة خسارة التنبؤ التراجعية الذاتية وقيود نسبة الضغط، مما يُمكّن النموذج من التحكم بفعالية في التكاليف الحسابية مع الحفاظ على دقة التنبؤ.
خلال مرحلة الاستدلال، يحدد النموذج ديناميكيًا طريقة تقسيم الكتلة بناءً على احتمالية الحدود، بحيث يمكن لدقة النمذجة أن تتكيف مع السياق وبالتالي تشبه بنية الجينوم الحقيقية بشكل أوثق.
يقلل برنامج dnaHNet التكلفة الحسابية بمقدار 3.89 مرة ويتفوق على برامج الأداء الأخرى متعددة المهام.
لتقييم أداء dnaHNet بشكل منهجي، قارنت هذه الدراسة بينه وبين نموذجين رئيسيين للجينوم ذي التسلسل الطويل: StripedHyena2 و Transformer++.تغطي التجارب جوانب متعددة، بما في ذلك خصائص القياس، والتنبؤ بتأثير التباين في العينة الصفرية، والتنبؤ بضرورة الجينات، ونمذجة البنية البيولوجية.
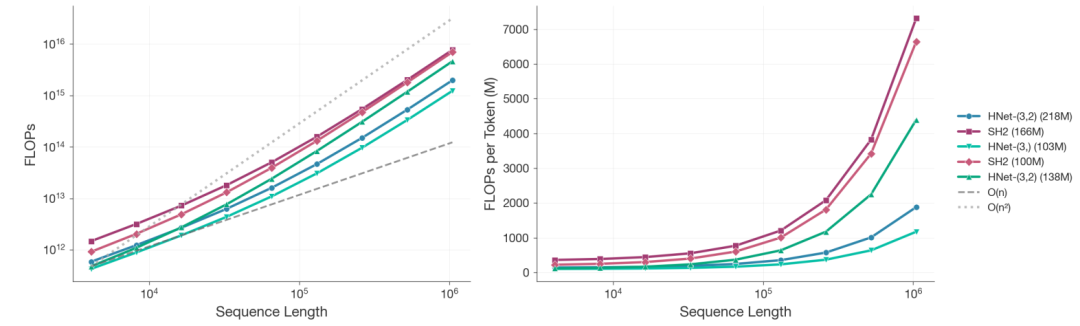
في تحليل القياس،قام الباحثون بتدريب أكثر من 100 نموذج بأحجام مختلفة ضمن ميزانية حوسبة ثابتة. عندما وصل طول التسلسل إلى 10⁶ نيوكليوتيدات وبلغت قوة الحوسبة الإجمالية 8 × 10¹⁹ عملية حسابية،تم تقليل التكلفة الحسابية لـ dnaHNet مع 218 مليون معلمة بمقدار 3.89 مرة تقريبًا مقارنة بـ StripedHyena2 مع 166 مليون معلمة.إن الهيكل ذو المرحلتين أكثر كفاءة من النسخة ذات المرحلة الواحدة.
تُظهر نتائج مطابقة قانون القوة بناءً على الحيرة أن،لتحقيق نفس مستوى الأداء، يتطلب StripedHyena2 قوة حاسوبية تبلغ حوالي 3.75 ضعف قوة dnaHNet.علاوة على ذلك، فإن التكوين الأمثل لمعلمات البيانات في dnaHNet ينحرف بشكل كبير عن قانون القياس التقليدي: ففي ظل نفس قوة الحوسبة، يمكن أن يصل عدد رموز التدريب الخاصة به إلى 140 مليار، بينما يحتوي نموذج المقارنة على 68 مليار فقط ولم يتقارب بعد.
في المهام اللاحقة،يتفوق dnaHNet باستمرار على نماذج المقارنة في كل من التنبؤ بتأثير تباين البروتين بدون عينة (MaveDB) والتنبؤ بضرورة الجينات (DEG).علاوة على ذلك، تتسع مزاياها مع زيادة القدرة الحاسوبية. وهذا يشير إلى أن آليتها الديناميكية القائمة على الكتل وبنيتها الهرمية تُمكنها من دمج بناء الجملة المحلي للترميز والمعلومات السياقية العالمية بشكل أكثر فعالية، مما يعزز قدرتها على توصيف الوظائف البيولوجية.
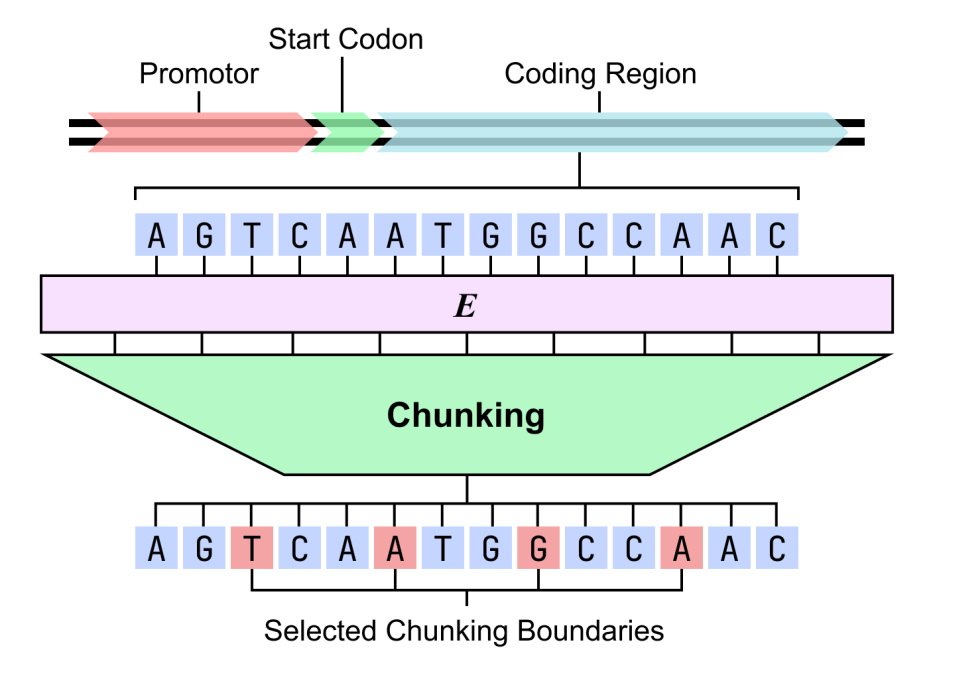
فيما يتعلق بتفسير البنية، تم تحليل جينوم بكتيريا Bacillus subtilis باستخدام نموذج dnaHNet ثنائي المراحل. أظهرت النتائج أن النموذج قادر على تعلم البنى الهرمية ذات الدلالة البيولوجية تلقائيًا: فقد أظهرت المرحلة الأولى حساسية تجاه الكودونات وقدرتها على رصد أنماط الثلاثيات بدقة في المناطق المشفرة؛ بينما ركزت المرحلة الثانية بشكل أكبر على البنى الوظيفية، حيث أظهرت المحفزات وكودونات البدء والمناطق بين الجينات احتمالات تجزئة أعلى بكثير من المناطق المشفرة.
تشير هذه النتيجة إلى أنلا يمتلك النموذج قدرات تنبؤية عالية الأداء فحسب، بل يمكنه أيضًا إعادة بناء التنظيم الوظيفي للجينوم في ظل ظروف غير خاضعة للإشراف.يوفر هذا مسارًا حسابيًا قابلًا للتفسير لتحليل "قواعد الحمض النووي".
خاتمة
بشكل عام، لم يعد نموذج dnaHNet يُحدد مسبقًا أساليب تجزئة التسلسل، بل يسمح للنموذج بتعلمها تلقائيًا. تُظهر التجارب أن هذا النموذج الديناميكي الهرمي لا يُحسّن الكفاءة الحسابية فحسب، بل يُعكس أيضًا بنية الجينوم متعددة المستويات بشكل أفضل. على المدى البعيد، إذا تمكن من تعلم الوحدات البيولوجية ذات الدلالة بثبات، فإنه يُبشّر بالكشف عن أنماط يصعب صياغتها في الجينوم، مما يفتح آفاقًا جديدة للبحث في التنبؤ بالتغيرات، والاكتشاف الوظيفي، والتصميم التركيبي.