Command Palette
Search for a command to run...
ICML 2025に選出された清華大学/中国人民大学/バイトダンスは、多種類の薬物分子設計を実現する初の分子間統合生成フレームワークUniMoMoを提案した。

清華大学の劉洋教授率いるグループ、中国人民大学高陵人工知能学院の黄文兵教授率いるグループ、およびByteDance AI製薬チームは共同で、分子種間の統一された生成フレームワークであるUniMoMoを提案した。このフレームワークは、分子フラグメント (ブロック) に基づいてさまざまな種類の分子を均一に表現し、変分オートエンコーダを使用して各ブロックの完全な原子構造を圧縮し、圧縮された潜在空間で幾何学的拡散モデリングを実行することで、同じターゲットに対する異なる種類の結合分子 (小分子、ペプチド、抗体) の設計を実現します。 UniMoMo は、複数の分子タスクベンチマークでトップクラスのパフォーマンスを実現し、クロスモーダルな知識転送とデータ共有の大きな可能性を実証しています。

関連する成果は、「UniMoMo: De Novoバインダー設計のための3D分子の統合生成モデリング」というタイトルでICML 2025に選出されました。
用紙のアドレス:
オープンソース プロジェクトのアドレス:
https://github.com/kxz18/UniMoMo
統合モデリングの理由
医薬品開発においては、分子の種類ごとに長所と短所があるため、さまざまな疾患シナリオで最も適切な分子の種類を選択する必要があることがよくあります。例えば:
* 小分子はサイズが小さく、経口摂取しやすく、浸透性が強いため、細胞に入り込んで標的に作用するのに適しています。これらは慢性疾患や代謝性疾患に広く使用されています。
* ペプチド分子は標的化特性が高く、タンパク質表面の広く平坦な領域に結合することができます。これらは、「薬物治療が難しい」タンパク質相互作用部位を標的とするのに適しており、がんや炎症などの治療によく使用されます。
* 抗体は選択性と親和性が極めて高く、特定のタンパク質マーカーを安定的に識別できるため、免疫療法などの精密な介入シナリオに特に適しています。
したがって、疾患のメカニズム、標的の特性、薬物治療のニーズが異なると、使用に適した分子の種類も異なります。既存の生成方法では通常、特定のクラスの分子(小分子、ペプチド、抗体など)のみをモデル化します。多様な治療ニーズを満たすことも、異なる分子間の共通性を活用してモデルのパフォーマンスを向上させることもできません。
アプリケーションの観点から見ると、統合モデリングにより、同じターゲットに対して複数の種類の薬剤候補を同時に探索することができ、さまざまな下流シナリオにさらに多くのオプションを提供できます。
機械学習の観点から見ると、異なる種類の分子は類似した結合規則(水素結合、π-πスタッキング、塩橋など)と幾何学的制約(結合長、結合角など)を共有しており、お互いから学習することができます。したがって、統合モデリングでは、より大きなデータ規模を活用して、モデルの一般化と相互転送機能を向上させることができるはずです。
生成的統一モデリングの難しさ
異なるタイプの分子を均一に生成するというアイデアは魅力的ですが、そのようなフレームワークを実現するには、主に分子表現の選択と生成アルゴリズムの設計において、依然として大きな課題が残っています。
まず、異なる分子タイプの構造表現には大きな違いがあります。小さな分子はさまざまな官能基で構成されており、その構造は非常に多様で非線形です。一方、ペプチドや抗体はアミノ酸が直線的に連結して構成されており、特に抗体は明確な機能領域の区分を持っています。直感的ではあるがよくないアプローチは、すべての分子を原子のグラフとしてモデル化することです。しかし、このアプローチでは、ベンゼン環や標準アミノ酸などの主要な部分構造など、分子の自然な階層構造が無視されるため、抗体などの大きな結合面を持つシステムを扱う際に計算コストが非常に高くなります。
逆に、フラグメントレベルのグラフを構築するために共通の構造フラグメント語彙のみを使用する場合(例えば、ほとんどのタンパク質設計作業ではCα 座標)、原子レベルの詳細を無視すると、分子生成の移植性と精度が犠牲になります。結合分子の設計における基本的な法則は、標的との空間的な相互作用と分子内の幾何学的制約であるため、これらは原子レベルで定義される物理法則であり、正確な全原子情報のサポートが必要です。
したがって、真に効果的かつ効率的な統一された分子表現を構築するには、次の 2 つの課題を同時に解決する必要があります。構造的な階層的事前条件を抽象化しながら、原子レベルで幾何学的詳細を保持する必要があります。
第二に、階層的な事前確率を保持するために構造フラグメントが生成に導入されると、生成アルゴリズムに中核的な課題が生じます。従来の拡散モデルは通常、固定長、固定構造のデータ表現に依存します。たとえば、固定数の点群または原子。 AF3 などの構造予測モデルでは、2D トポロジーが事前に与えられているため、拡散プロセスによって原子数や 2D 構造が変化することはありません。分子生成のタスクでは、2D トポロジと 3D 構造を同時に生成する必要があります。ノイズ除去プロセス中に構造フラグメントの種類が変化すると、それに応じて原子の対応する数、種類、配置も変化します。これにより、従来の拡散モデルの前提が崩れ、モデリングに極めて高い要求が課せられます。
UniMoMo: 統合生成モデル
異なる分子タイプ間の構造の違いが大きく、モデリングが困難であるという問題を解決するために、この記事では新しいフレームワーク UniMoMo を提案しています。それは、構造階層と原子レベルの精度を効果的に考慮した 2 つの主要な設計から始まります。
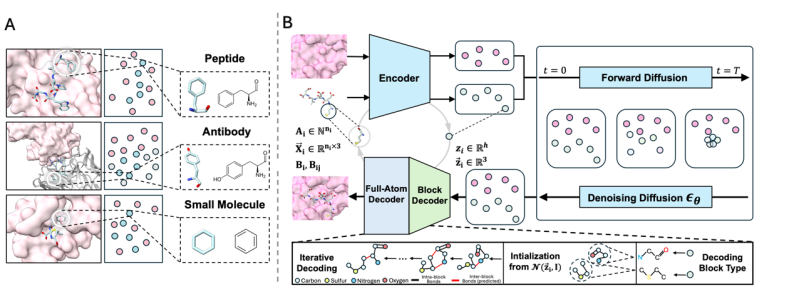
* 統一表現:すべての分子タイプはブロック形式でモデル化されます。
UniMoMo は、小分子、ペプチド、抗体など、その構造を分子フラグメント (ブロック) で構成されたグラフとして表現します。各ブロックは、標準アミノ酸または一般的な小分子フラグメント(ベンゼン環、インドールなど)になります。この記事の実装では、記録された分子フラグメントには、すべての標準アミノ酸と、主要なサブグラフマイニングアルゴリズムによって自動的に識別された小分子フラグメントが含まれます。すべての非天然アミノ酸は小分子として分類できます。この表現では、分子の原子レベルの詳細と、さまざまな種類の分子自体の階層構造の両方が保持されるため、統一されたモデリングが可能になります。
* 全原子幾何学的暗黙空間拡散モデル:圧縮された表現での効率的な生成。
生成過程におけるブロックタイプの変化によって原子の種類と量が同期的に変化する問題を解決し、生成効率と構造精度を向上させるために、この記事では、全原子反復変分オートエンコーダ (IterVAE) を設計します。各ブロック内のすべての原子は、固定長の潜在空間表現ベクトルと対応する潜在空間座標を含む潜在空間内の「ポイント」に圧縮されます。
次に、モデルは圧縮された幾何学的潜在空間で生成モデリングを実行し、新しい分子の潜在表現を生成し、最終的に完全な原子構造にデコードされます。潜在空間のデータ表現は固定長(ブロック数が事前に与えられている)かつ連続的であるため、既存のさまざまな生成アルゴリズムと簡単に互換性を持たせることができます。現在の試みでは、拡散モデルは比較的良好な結果を生み出すことができています。この設計により、モデルは生成プロセス中にブロック間のグローバルレイアウトに焦点を当てることができ、詳細な原子レベルの構造はデコーダーによって完成されるため、高い効率と原子レベルの精度の統一が達成されます。
統合モデリングは単一ドメインモデリングを超える
さまざまな種類の分子に対する UniMoMo の汎用性と有効性を検証するために、著者らは複数の構造ベースの設計タスクで体系的な評価を実施しました。これは、小分子、ペプチド、抗体という 3 つの代表的な結合分子の種類をカバーしています。本実験は、当該分野における最も代表的な単分子型生成モデルと比較することにより、特に空間構造の合理性や結合能力などの重要な指標に関して、統合モデリングがより強力な幾何学的モデリング能力とクロスモーダル一般化能力を有するかどうかを探ることを目的とします。
結果は次のようになります。UniMoMoは均一に訓練され、すべての分子タイプにおいて総合的な優位性を獲得しました。構造修復精度に優れているだけでなく、主要な幾何学的合理性やターゲットとの相互作用の品質も大幅に向上します。
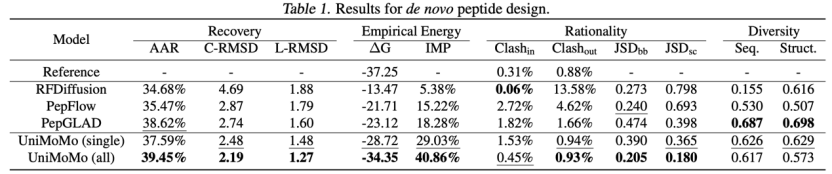
ペプチド生成タスクでは、UniMoMo は、複数の主要指標において既存のドメイン固有モデルを大幅に上回ります。RFDiffusion、PepFlow、PepGLAD などを含みます。特に構造精度の点では、UniMoMo は複合体とモノマーの RMSD が低く、生成されたペプチド構造が実際の結合構造に近いことを示しています。
UniMoMo は、Rosetta 結合エネルギーが低い構造も生成できます。これは、タンパク質結合部位の幾何学的特徴に対するより強力なモデリング能力を反映しています。さらに、UniMoMo は、ペプチド構造の品質を測定する二面角分布の一貫性 (バックボーン/側鎖のねじれの JSD) や原子レベルの空間衝突 (衝突率) などの幾何学的合理性指標でもトップクラスのパフォーマンスを示しました。さらに、すべてのデータを使用してトレーニングされた UniMoMo (all) は、さまざまな指標において、ペプチド データのみを使用してトレーニングされたモデルよりも一貫して優れたパフォーマンスを発揮しました。UniMoMo が分子種にわたって学習し、一般化する能力が実証されています。
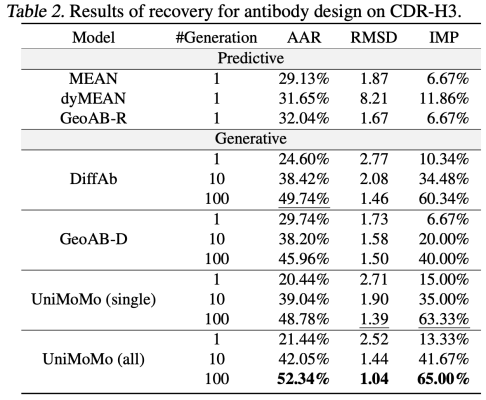
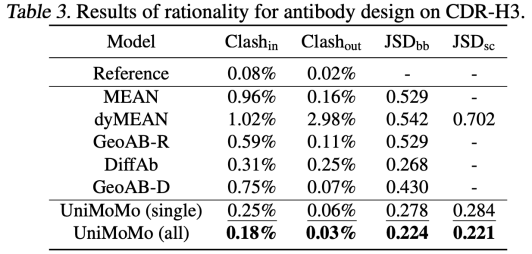
UniMoMo は抗体設計タスクでも優れたパフォーマンスを発揮しました。 MEAN、dyMEAN、DiffAbなどの既存の方法と比較して、UniMoMo は、自然に結合した配列と構造の再現率 (AAR と RMSD) や結合エネルギーの改善 (IMP) などの主要な指標に関して、他のすべてのターゲットを上回っています。特に、多重サンプリング生成の評価において、UniMoMo はより高い確率で天然構造に近い抗体断片を生成することができ、抗体構造空間における優れた探索能力を示しています。
同様に、異なる分子タイプのデータを使用して共同でトレーニングされた UniMoMo(all) は、すべての指標において抗体データのみを使用してトレーニングされたバージョンよりも優れています。これは、統一されたモデリングが、モデルが分子構造のより普遍的で転送可能な空間法則を学習するのに役立つことを示しています。この結果は、異なる分子タイプ間の構造モデリングの共通性を強調し、生成品質の向上におけるクロスドメインデータ融合の重要な価値を検証します。
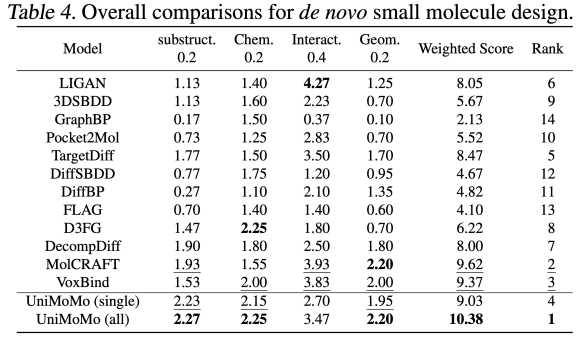
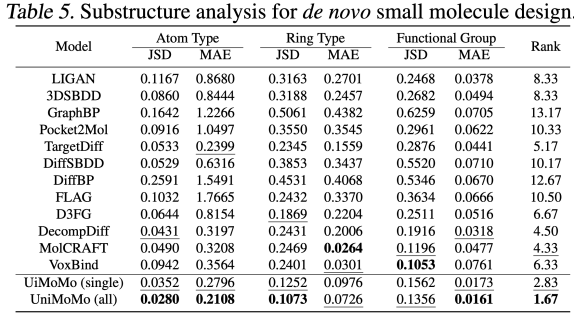
UniMoMo は、小分子生成タスクでも優れたパフォーマンスを発揮しました。 CrossDocked2020データセットの評価を通じて、著者らは、CBGBench に基づく総合評価において、UniMoMo が既存の主流の手法を上回っていることを発見しました。
具体的には、UniMoMoは、部分構造分布(原子種、官能基など)、化学的性質の合理性(QED、LogP、SAなど)、幾何構造品質(結合長・角度分布、原子衝突率など)、相互作用スコア(Vinaドッキング)において高い総合スコアを達成しました(完全な実験結果については原文をご参照ください)。重要なのは、分子タイプ全体でトレーニングされた UniMoMo(all) は、小分子データのみでトレーニングされた単一ドメイン バージョンと比較して、すべての評価次元で大幅な改善を示していることです。これは、最も柔軟な分子構造と最も多様なタイプを持つ小分子シナリオでも、統一モデルは他の分子タイプから幾何学的法則と相互作用パターンを転送できるため、小分子のモノマー構造と相対的なポケット空間レイアウトの合理性が向上します。この現象は、UniMoMo の中核概念を改めて証明しています。つまり、異なる分子間の幾何学的制約と結合メカニズムには共有可能なパターンがあり、統一されたモデリングによってこの可能性を効果的に刺激できるということです。
3 種類のタスクの実験結果を組み合わせると、UniMoMo は非常に一貫した利点を示しています。分子種間データを使用してトレーニングされた統合モデルは、それぞれのタスクで既存の単一ドメイン生成モデルよりも優れており、単一ドメイン データのみでトレーニングされた UniMoMo と比較して明らかに機能が向上しています。この現象は、分子設計における一見異なるタスクが、実際には根底にある物理的および化学的制約と空間幾何学的法則において高度な共通性を持っていることを示しています。UniMoMo の統合モデリング戦略は、この共通性を捉えて増幅し、それによってタスク間の転送と補完的な強化を実現します。これらの結果は、UniMoMo の有効性を検証するだけでなく、将来的にさらに強力な統合分子生成システムを構築するための強力な経験的サポートも提供します。
GPCRのケーススタディ
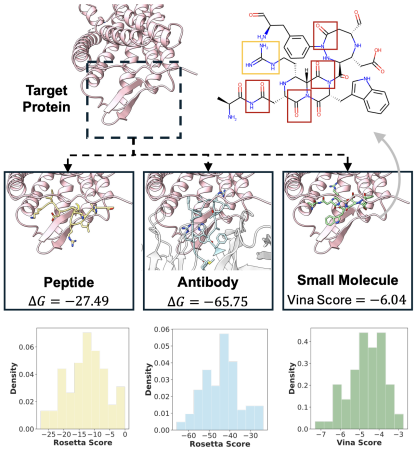
ケーススタディとして、著者らはヒトにおける最も重要な薬物ターゲットの 1 つである G タンパク質共役受容体 (GPCR) を選択し、同じ結合部位で異なる種類の分子 (ペプチド、抗体、小分子) を生成する UniMoMo の能力を評価しました。 UniMoMo によって生成されたペプチド、抗体、小分子はすべて、結合エネルギー評価に一般的に使用される力場 (Rosetta ΔG、Vina スコア) 下で良好な分布を示します。さらに驚くべきことは、生成された小分子構造が、水素結合を構築し、標的との重要な相互作用を形成するために使用される天然アミノ酸側鎖に類似した官能基も自発的にシミュレートすることです。さらに、小分子は分子骨格上のアミド結合などの局所的な幾何学的構成をペプチドや抗体から借用しており、これにより、本来は大きな分子に適している結合ポケットを効果的に満たすことができます。この事例は、UniMoMo がモダリティ間で借用し、実際のタスクの結合ポケットに自動的に適応する能力を鮮明に示しており、ターゲットと分子間の相互作用と分子の内部の幾何学的制約を 3 次元構造レベルで深く理解する可能性を反映しています。
将来の探査
UniMoMo は複数の分子タイプとタスクで強力な統合生成機能を実証していますが、著者らは、この方向で探求する価値のある将来の可能性がまだたくさんあることも指摘しました。
現在の研究は主に天然アミノ酸と一般的な分子フラグメントのモデリングに焦点を当てており、これは非天然アミノ酸、後修飾ペプチド/抗体、環状分子などのより複雑な薬物形態にさらに拡張することができ、より広範囲の候補分子空間をカバーします。統一モデリングの概念は、モデルの制御可能性と解釈可能性を研究する機会も提供し、生成モデルをさらに信頼性が高く実用的な分子設計プラットフォームへと発展させることが期待されます。要約すると、UniMoMo の導入により、分子設計タスクのための汎用的かつ強力な生成フレームワークが提供されるだけでなく、AI 主導の創薬の可能性に満ちた新しい方向性も開かれます。








