Command Palette
Search for a command to run...
L'IA Appliquée Aux Matériaux Évolue Vers Une « Ère Explicable » : Une Équipe Japonaise a Percé Le Mystère De La Spectroscopie Haute Dimensionnelle, Identifiant Des Caractéristiques Clés Pour La Découverte De Nouveaux matériaux.

Ces dernières années, l'application de l'apprentissage automatique en science des matériaux a suscité un vif intérêt. Son champ d'application s'est progressivement étendu, passant de la prédiction de relations structure-propriété (telles que l'énergie de bande interdite, l'énergie de formation des défauts ponctuels, le point de fusion, etc.) à la modélisation de grandeurs physiques multidimensionnelles plus complexes. L'un des axes de recherche les plus prometteurs concerne la prédiction et l'analyse des spectres de matériaux.
Les données spectrales, telles que les fonctions diélectriques, les spectres (absorption, réflexion et émission) et les densités d'états électroniques et phononiques, sont essentielles à la compréhension et à la conception des matériaux. Cependant, contrairement aux propriétés scalaires, les données spectrales de haute dimensionnalité se caractérisent par des dimensions de sortie importantes, des structures complexes et de fortes contraintes physiques, ce qui rend difficile pour les méthodes d'apprentissage automatique traditionnelles d'atteindre simultanément précision et interprétabilité. Bien que les modèles d'apprentissage profond aient été capables de prédire les spectres dans une certaine mesure, le manque d'interprétabilité demeure un obstacle majeur limitant leur application à la conception des matériaux.
Dans ce contexte,Une équipe de recherche de l'Institut des sciences de Tokyo, au Japon, a proposé une méthode d'interprétation des modèles d'apprentissage profond capables de traiter des données spectrales de haute dimension en science des matériaux.Des chercheurs ont constitué un ensemble de données à partir de calculs ab initio des spectres d'absorption optique de 2 681 oxydes, chalcogénures et composés apparentés. Après correction de l'énergie et de la forme du seuil d'absorption, les résultats obtenus présentent une concordance nettement améliorée avec les spectres expérimentaux publiés, comparativement aux calculs classiques basés sur la théorie de la fonctionnelle de la densité.
Les chercheurs ont également utilisé l'ensemble de données et l'algorithme ALIGNN pour développer un modèle de prédiction du spectre d'absorption optique de haute précision.En combinant l'extraction de caractéristiques et l'analyse de regroupement, les types d'éléments clés et leurs environnements de coordination qui déterminent principalement l'énergie et l'intensité d'initiation de l'absorption de la lumière ont été extraits avec succès.
Les résultats de recherche connexes, intitulés « Extraction par apprentissage profond de groupes de matériaux prometteurs et de caractéristiques communes à partir de données de grande dimension : le cas des spectres optiques de cristaux inorganiques », ont été publiés dans Advanced Intelligent Discovery.
Points saillants de la recherche :
Cette étude propose une méthode de classification des matériaux par extraction de caractéristiques et analyse de regroupement de données spectrales de grande dimension, permettant ainsi d'extraire des groupes de matériaux potentiels et leurs caractéristiques communes.
* L'ensemble de données de calcul ab initio et le modèle d'apprentissage automatique développés dans cette étude devraient jouer un rôle important dans la découverte future de matériaux et la recherche en informatique des matériaux.
La méthode proposée dans cette étude présente une large applicabilité et peut être utilisée pour la classification et l'interprétation de diverses données spectrales. Son application ne se limite pas aux spectres d'absorption optique des cristaux inorganiques.

Adresse du document :https://advanced.onlinelibrary.wiley.com/doi/10.1002/aidi.202600007
Ensembles de données construits à l'aide de calculs ab initio à haut débit
Les chercheurs ont d'abord sélectionné, dans la base de données Materials Project, les oxydes, les chalcogénures et les matériaux apparentés répondant aux critères suivants : (1) le matériau contient au moins un des éléments O, S et Se, et son degré d'oxydation n'est pas nécessairement −2 ; (2) le matériau ne contient pas les éléments suivants : H, halogènes, gaz rares, Mn–Ni, Tc–Rh, Os–Ir, Po, lanthanides (à l'exception de La et Ce) et actinides ; (3) le matériau ne présente pas de polarisation de spin ; (4) les systèmes de groupe d'espace P1 et/ou comportant plus de 40 atomes dans la maille élémentaire d'origine ont été exclus en raison d'un coût de calcul élevé ou d'une incertitude quant à leur structure cristalline.
Le nombre total de matériaux utilisés dans les calculs ab initio était de 9 808, et la base de données de calcul a été construite selon le processus illustré dans la figure ci-dessous.
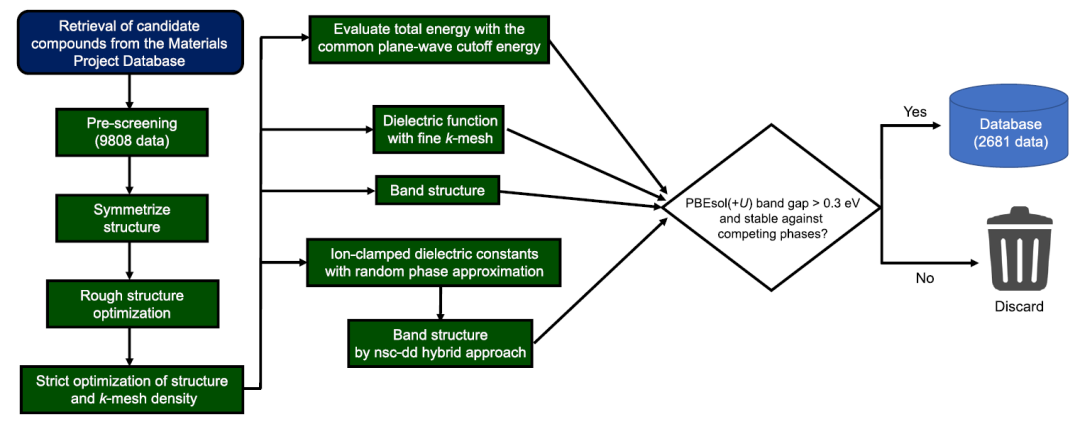
Comme le montre la figure, ce processus de calcul est extrêmement complexe. Afin d'obtenir un calcul à haut débit tout en garantissant la cohérence et l'utilisation efficace des ressources de calcul,Les chercheurs ont utilisé leur propre programme développé et se sont appuyés sur des outils tels que pymatgen, FireWorks, Custodian, atomate et vise pour automatiser le processus.Tous les calculs ab initio ont été effectués à l'aide du logiciel VASP. Ce flux de travail utilise des calculs PBEsol(+U) pour générer les spectres d'absorption optique et les énergies de formation des composés, et des fonctionnelles mixtes nsc-dd et des calculs PBEsol(+U) pour obtenir la structure de bandes.
Concernant l’ensemble de données d’apprentissage automatique, les chercheurs ont éliminé : (1) les matériaux instables par rapport à la phase concurrente dans la base de données locale ; et (2) les matériaux dont la bande interdite PBEsol(+U) est inférieure à 0,3 eV. Le nombre final de matériaux retenus était de 2 681.
Construction d'un modèle ALIGNN basé sur les spectres d'absorption optique
construction de modèles d'apprentissage automatique et précision des prédictions
Au niveau du modèle,Cette étude utilise ALIGNN (Atomistic Line Graph Neural Network) comme cadre de prédiction principal pour modéliser les spectres d'absorption optique de haute dimension.Comparé aux réseaux de neurones convolutifs à graphes cristallins (CGCNN) traditionnels, l'avantage principal d'ALIGNN réside dans l'introduction simultanée d'une double représentation « graphe atomique + graphe de liaisons », encodant ainsi explicitement les informations relatives aux angles à trois corps et permettant une expression plus fine de l'environnement structural local. La partie supérieure de la figure ci-dessous présente un schéma de l'architecture d'ALIGNN.
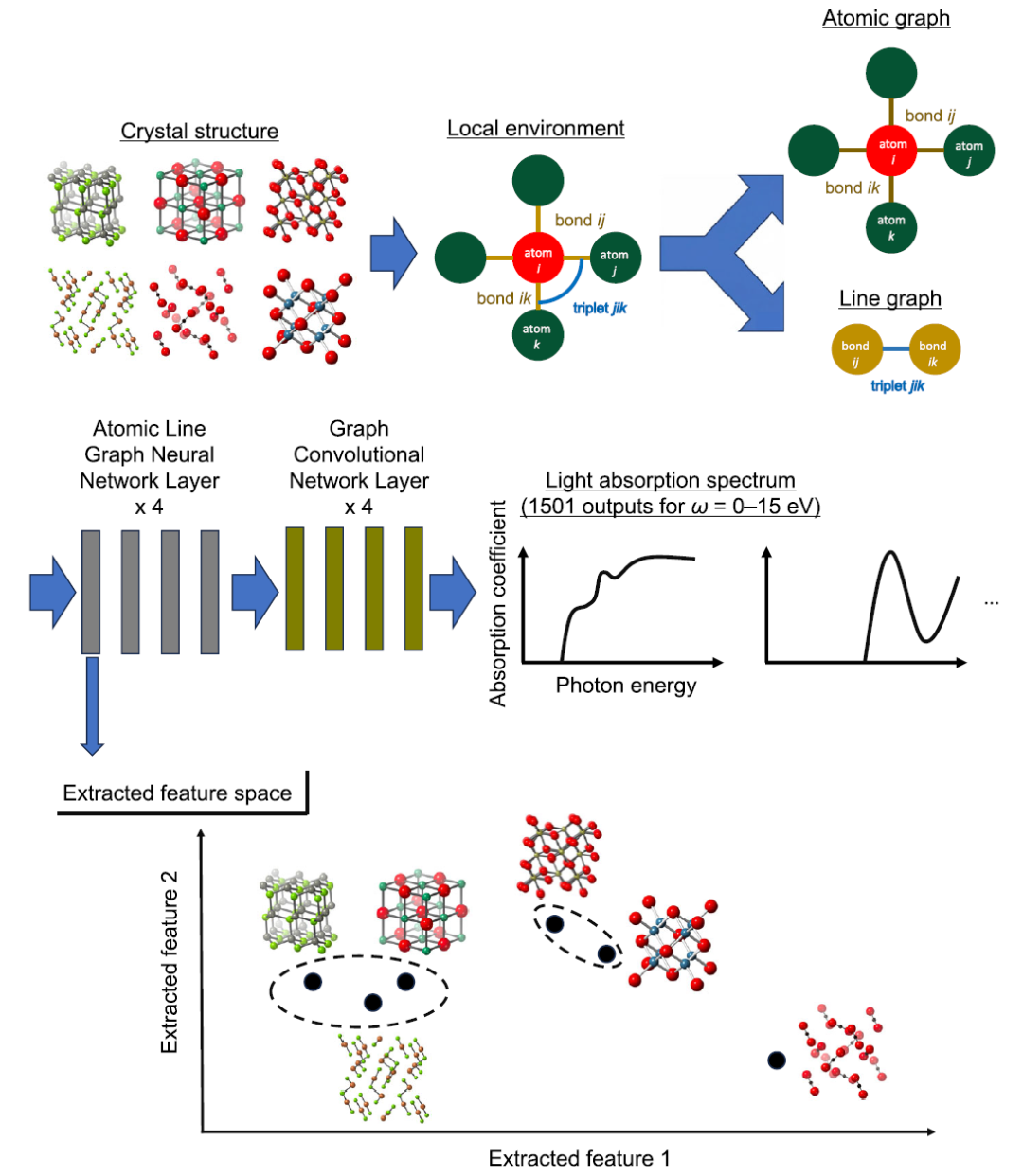
Dans ce cadre, les atomes sont des nœuds, les liaisons interatomiques sont des arêtes, et les relations entre les arêtes sont ensuite construites sous forme de graphes linéaires, transformant ainsi les informations sur les angles de liaison en caractéristiques structurelles apprenables.Cette conception permet au modèle non seulement de saisir les informations de distance entre deux corps, mais aussi de caractériser l'interaction entre trois corps, ressemblant ainsi davantage au comportement physique des cristaux.
Extraction de caractéristiques et regroupement
Les chercheurs ont extrait les caractéristiques de la première couche du modèle ALIGNN optimisé et ont calculé la moyenne des vecteurs de caractéristiques de tous les sites atomiques pour chaque matériau avant de procéder à une analyse de regroupement hiérarchique, comme illustré dans la partie inférieure de la figure ci-dessus. Cette méthode vise à classer les matériaux en groupes présentant des similarités tant au niveau des caractéristiques d'entrée (telles que la composition élémentaire et les caractéristiques de coordination atomique, notamment le nombre d'atomes adjacents, les distances interatomiques et les angles de liaison) que des propriétés de sortie (spectres d'absorption optique).
La figure ci-dessous présente les spectres d'absorption optique des 96 groupes obtenus par classification hiérarchique. La similarité des profils spectraux au sein de chaque groupe confirme l'efficacité de la méthode de classification employée dans cette étude.

Résultats : Une extraction interprétable de la structure de la population matérielle et des mécanismes physiques a été réalisée.
Pour vérifier la capacité du nouveau modèle d'apprentissage profond à traiter des données spectrales de haute dimension en science des matériaux, les chercheurs ont mené une série d'expériences :
Capacité de prédiction des performances
En termes de performances de prédiction, le modèle ALIGNN a démontré une précision globale élevée sur l'ensemble de test, comme le montre la figure ci-dessous.L'erreur absolue moyenne (MAE) de la prédiction du spectre d'absorption du matériau pour environ 75% est inférieure à 0,14, ce qui indique que le modèle peut bien reproduire des formes spectrales complexes.
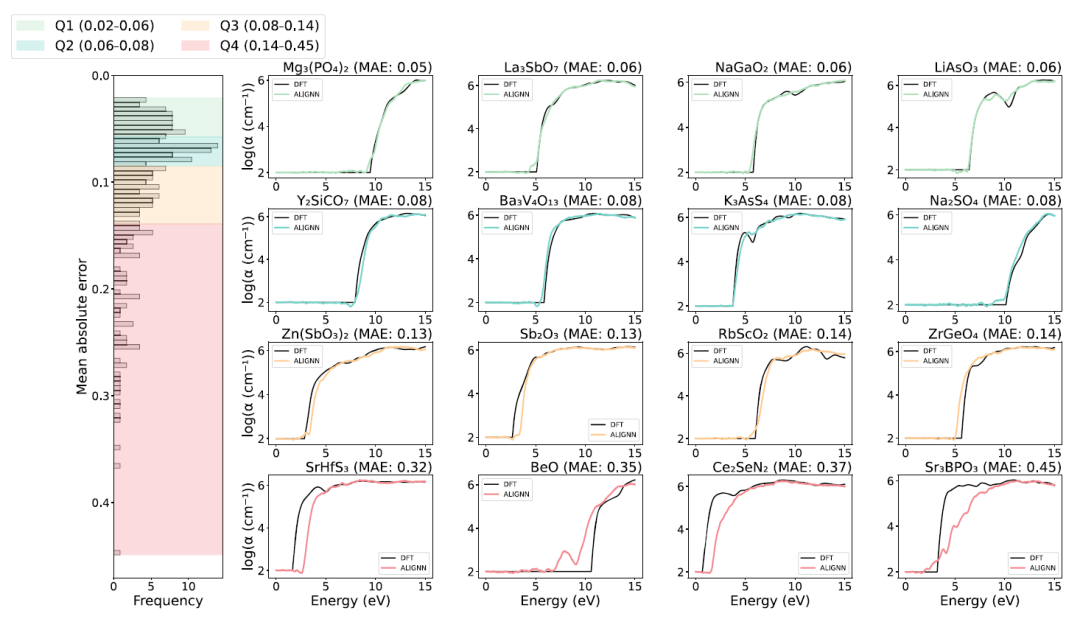
Le panneau de droite de l'image ci-dessus présente les résultats de prédiction pour les quatre matériaux présentant les erreurs les plus importantes dans chaque quartile. Pour les matériaux des trois premiers quartiles, les résultats de prédiction d'ALIGNN (courbes colorées) concordent bien avec les résultats de calculs de référence ab initio (courbes noires). Cependant, certains composés du quatrième quartile présentent des écarts significatifs dans la position initiale de leurs spectres d'absorption optique. Ces échantillons aberrants affichent de faibles performances de prédiction, principalement en raison de leurs structures électroniques uniques et de l'absence de matériaux de structure similaire dans l'ensemble de données d'entraînement.
La capacité à capturer la position initiale des spectres d'absorption optique
Bien que l'erreur absolue moyenne (MAE) soit une mesure globale couvrant l'ensemble du spectre, les chercheurs ont examiné plus en détail si le modèle pouvait reproduire avec précision l'énergie d'initiation spectrale locale. La figure ci-dessous présente un diagramme de parité : elle compare l'énergie photonique minimale correspondant au moment où log₁₀ α(ω) dépasse 2,5 dans les calculs ab initio et les prédictions d'ALIGNN, où α représente le coefficient d'absorption.
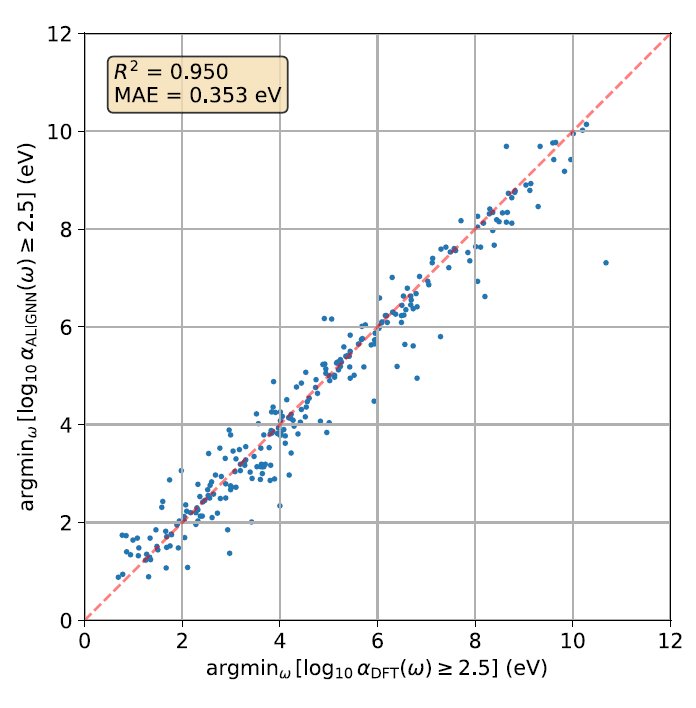
Les résultats montrent que le R² prédit pour l'énergie initiale est de 0,950 et que l'erreur absolue moyenne (MAE) est de 0,353 eV, ce qui indique que le modèle ALIGNN peut capturer avec précision la position initiale du spectre d'absorption optique.
Analyse d'interprétabilité
En termes d'analyse d'interprétabilité, les chercheurs ont extrait des représentations de caractéristiques de la première couche d'ALIGNN et effectué un regroupement hiérarchique des matériaux, aboutissant à 96 groupes de matériaux. Les résultats montrent que…Les matériaux appartenant à un même groupe présentent une grande homogénéité spectrale, notamment en ce qui concerne la position d'initiation de l'absorption et la pente du seuil d'absorption, ce qui témoigne d'une forte similarité. Ceci indique que le modèle a intégré les caractéristiques structurales spectrales des premières couches.
Des études de cas supplémentaires révèlent des différences physiques nettes entre les différents groupes de matériaux. Par exemple, le groupe 74 présente généralement de larges bandes interdites et des coefficients d'absorption élevés au voisinage du point d'inflexion spectral. La figure a montre que tous les matériaux de ce groupe contiennent soit du vanadium (V), soit du chrome (Cr), les autres cations étant principalement des métaux alcalins. Ces matériaux existent le plus souvent sous les formes VO₄³⁻, CrO₄²⁻ ou Cr₂O₇²⁻, les cations étant situés dans un environnement de coordination tétraédrique.
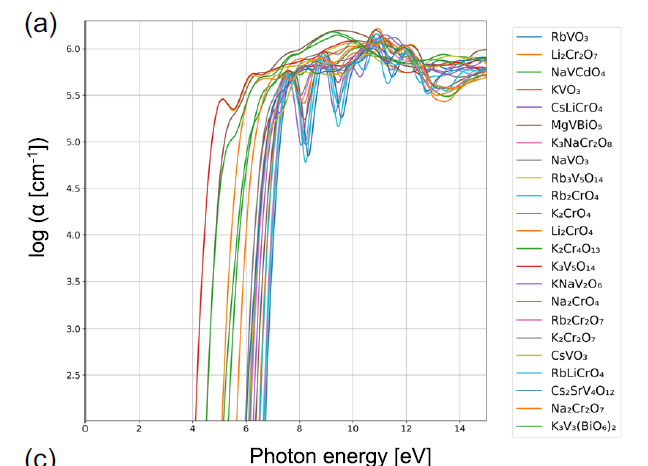
Spectres d'absorption optique des substances appartenant au groupe 74, où α représente le coefficient d'absorption.
Les chercheurs ont utilisé CrystalFingerprintNN, implémenté dans Matminer, pour calculer l'indice de coordination tétraédrique des sites cationiques dans chaque matériau du cluster et ont analysé la distribution des valeurs maximales pour tous les sites cationiques. Comme le montre la figure b ci-dessous, la plupart des matériaux possèdent effectivement des sites de coordination tétraédrique.
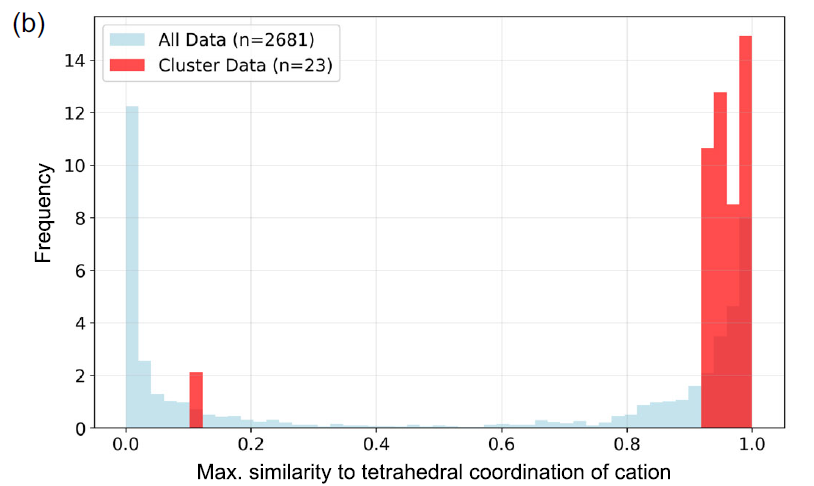
Distribution de la similarité de coordination tétraédrique entre le 74e groupe de matériaux (rouge) et l'ensemble des données (bleu).
Du point de vue de la densité d'états électroniques, un pic marqué, dû aux états d du vanadium (Vd) ou du chrome (Cr), est observable près du bas de la bande de conduction (BBC). Les états de valence élevés du V⁵⁺ et du Cr⁶⁺ offrent un grand nombre d'états électroniques inoccupés, susceptibles d'être utilisés pour des transitions optiques. Par conséquent, du point de vue de la chimie et de la physique du solide, il est logique que ces vanadates, chromates et dichromates présentent des coefficients d'absorption optique élevés.
Ce processus d'inférence des mécanismes chimiques à partir des résultats de regroupement de modèles transforme les prédictions d'apprentissage automatique, initialement opaques, en une source précieuse de connaissances pour la conception de matériaux. De plus, l'étude a comparé les résultats d'un regroupement direct basé sur des données spectrales brutes. Elle a constaté que, bien qu'il puisse identifier des spectres similaires, il peinait à former des groupes de structures chimiques distincts, entraînant un mélange important de types de matériaux. Ceci démontre une fois de plus l'avantage de l'espace de caractéristiques ALIGNN pour obtenir une représentation cohérente de la relation structure-propriétés.
Conclusion
L'importance de cette étude réside non seulement dans la construction d'un modèle de prédiction de haute précision pour la spectroscopie d'absorption optique, mais surtout dans la proposition d'un cadre méthodologique combinant l'apprentissage profond et l'interprétation physique des matériaux. En associant le modèle ALIGNN à une analyse de regroupement hiérarchique, l'étude permet d'extraire les lois générales des matériaux à partir de données spectrales multidimensionnelles, permettant ainsi aux modèles d'apprentissage automatique non seulement de prédire les résultats, mais aussi de révéler la structure sous-jacente et les origines électroniques de ces résultats.
Idéalement, les effets des interactions électron-trou, du couplage électron-phonon et des défauts ponctuels devraient être pris en compte pour reproduire les caractéristiques spectrales des effets excitoniques, des transitions électroniques assistées par phonons et des défauts, respectivement. Cependant, les calculs spectraux ab initio à haut débit incluant ces effets sont trop coûteux en ressources de calcul, ce qui explique pourquoi cela n'a pas été possible dans cette étude. Néanmoins, grâce à l'intégration plus poussée de méthodes de calcul à N corps plus précises et de modèles d'apprentissage automatique, ce type de recherche devrait jouer un rôle plus central dans la découverte de nouveaux matériaux, faisant passer la conception des matériaux d'une approche empirique à une nouvelle étape intégrant des approches basées sur les données et sur les mécanismes.