Command Palette
Search for a command to run...
Publiée Dans La Revue Nature, Une Évaluation Épidémiologique Des Eaux Usées Basée Sur Le Séquençage Génétique Et l'apprentissage Automatique Peut Détecter Les Virus jusqu'à 4 Semaines Plus tôt.

Ces dernières années, la sécurité sanitaire mondiale a été confrontée à de graves défis. Cela est particulièrement vrai depuis le début de la pandémie de COVID-19. Son agent pathogène, le coronavirus du syndrome respiratoire aigu sévère 2 (SRAS-CoV-2), a continué d'évoluer, avec l'émergence de multiples variants dominants. Ces variants possèdent des capacités variables à infecter et à échapper aux réponses immunitaires, ce qui accroît considérablement la difficulté de la prévention et du contrôle des épidémies et la charge pesant sur les systèmes de santé.
Les tests de santé publique et le séquençage du génome du SRAS-CoV-2 sont des moyens importants pour détecter de manière exhaustive les variantes en circulation.Cependant, ce type de surveillance clinique dépend souvent fortement d’une grande quantité de ressources de laboratoire et nécessite que les individus participent activement au test.Il est difficile de suivre intégralement l'émergence et la propagation des variants du SARS-CoV-2. En particulier dans les régions où les ressources médicales sont relativement limitées ou où la volonté de se faire dépister est faible, la surveillance clinique est plus sujette aux biais de détection, ce qui crée des angles morts en matière de prévention et de contrôle.
En tant qu’approche complémentaire, l’épidémiologie basée sur les eaux usées (WBE) a joué un rôle important dans l’alerte aux épidémies depuis qu’elle a été proposée pour la première fois dans les années 1940 pour évaluer l’infection communautaire. WBE détecte et suit principalement la composition virale et les changements dynamiques en analysant les traces de virus excrétés par le corps humain dans les eaux usées.Par rapport à la surveillance clinique, la WBE peut refléter de manière objective et impartiale la situation d'infection du groupe dans la zone couverte sans s'appuyer sur des tests actifs individuels, permettre une alerte précoce et présente un rapport coût-efficacité significatif.
Cependant, les méthodes actuelles de surveillance des eaux usées (telles que Freyja et COJAC basées sur la régression linéaire) présentent encore des limites.La détection doit être basée sur le modèle mutationnel des variantes connues (telles que les séquences de référence dans les bases de données GISAID ou UshER),Si une nouvelle variante qui n’a pas été caractérisée ou incluse dans la littérature clinique apparaît, il est souvent difficile de l’identifier avec précision, ce qui limite dans une certaine mesure l’efficacité de détection du WBE.
Pour résoudre ce problème, une équipe de recherche de l’Université du Nevada à Las Vegas a proposé une méthode d’analyse multivariée appelée ICA-Var (Independent Component Analysis of Variants).La méthode est basée sur une conception de processus d’apprentissage automatique non supervisé et utilise l’analyse en composantes indépendantes (ICA) pour extraire les modèles de covariation et de mutation évoluant dans le temps à partir des données sur les eaux usées.Une détection plus précoce et plus précise des variantes est obtenue.
Grâce à cette méthode, l’équipe de recherche a détecté avec précision la variante Delta, la variante Omicron et la variante recombinante XBB entre fin 2021 et 2023. Cette méthode réaffirme non seulement l’efficacité de la surveillance des eaux usées pour l’alerte précoce de la prévention et du contrôle des épidémies, mais fournit également un nouvel outil pour suivre de manière exhaustive les mutations virales et la propagation en l’absence de surveillance clinique.
La recherche connexe a été publiée dans Nature Communications sous le titre « Détection précoce des variants émergents du SARS-CoV-2 dans les eaux usées grâce au séquençage du génome et à l'apprentissage automatique ».
Points saillants de la recherche :
* Cette méthode révèle la dynamique spatiotemporelle des mutations virales dans les zones urbaines et rurales, confirme la loi de transmission du virus des zones urbaines aux zones rurales et fournit un paradigme de détection de variantes efficace et peu coûteux pour les zones ayant un accès médical limité ou un manque de données de séquençage clinique.
* Par rapport à l'outil de référence actuel Freyja, la méthode d'analyse multivariée d'ICA-Var présente des avantages significatifs, et le temps de détection des variantes Delta, Omicron et des dernières variantes EG.5, HV.1, BA.2.86 est en moyenne de 1 à 4 semaines plus tôt

Adresse du document :
https://www.nature.com/articles/s41467-025-61280-5
Collecte de données à long terme et multipoints
Dans cette étude, les échantillons d’eaux usées utilisés dans l’expérience ont été collectés d’août 2021 à novembre 2023.3 659 échantillons d’eaux usées ont été collectés dans les zones urbaines et rurales du sud du Nevada.Après la collecte, les échantillons d’eaux usées seront placés sur glace sur place et conservés au réfrigérateur jusqu’au traitement, pour une durée de conservation ne dépassant pas 36 heures.
Au cours du processus d’extraction de l’acide nucléique,L'équipe de recherche a d'abord isolé les acides nucléiques d'échantillons d'eaux usées à l'aide du kit Promega Wizard Enviro Total Nucleic (réf. A2991), conformément aux exigences réglementaires. Elle a ensuite modifié le protocole Promega en lysant les eaux usées avec une solution de protéase et en utilisant des billes Macherey-Nagel NucleoMag (réf. 744970) pour lier les acides nucléiques libres. Pour les ARN supérieurs à 10 ng, l'équipe a utilisé le kit LunaScript RT SuperMix de New England BioLabs pour la synthèse du premier brin d'ADNc.
Construction et séquençage de bibliothèques de séquençage,L'équipe de recherche a utilisé le panneau CleanPlex SARS-CoV-2 FLEX de Paragon Genomics pour construire des bibliothèques de séquençage d'amplicons, qui ont ensuite été séquencées sur la plate-forme Illumina NextSeq 500 ou NextSeq 1000 à l'aide d'une cellule à flux de 300 cycles.
En termes de traitement des données de séquençage,L'équipe a d'abord utilisé le logiciel cutadapt (version 4.2) pour supprimer les séquences d'adaptateur Illumina des paires de lectures de séquençage. Elle a ensuite mappé ces paires de lectures au génome de référence du SARS-CoV-2 (NC_045512.2) à l'aide du logiciel bwa mem (version 0.7.17-r1188). Elle a ensuite utilisé l'outil fgbio TrimPrimers (version 2.1.0, mode de découpage strict) pour supprimer les séquences d'amorces d'amplicon Paragon Genomics CleanPlex SARS-CoV-2 FLEX des lectures alignées. Enfin, le logiciel iVar Variants (version 1.4.1) a été utilisé pour détecter les variants (sur la base des différences de fréquence des allèles par rapport au génome de référence original de 2020), et le logiciel samtools (version 1.16.1) pour calculer la couverture génomique et la profondeur de lecture.
Après avoir retiré les échantillons en double et les contrôles positifs/négatifs,Les 2 684 échantillons restants ont été utilisés pour l’analyse de contrôle qualité (CQ).Après un contrôle qualité rigoureux, seuls les échantillons d'eaux usées avec une profondeur de séquençage de 50x et couvrant au moins 80% du génome du SARS-CoV-2 ont été conservés pour une analyse ultérieure.
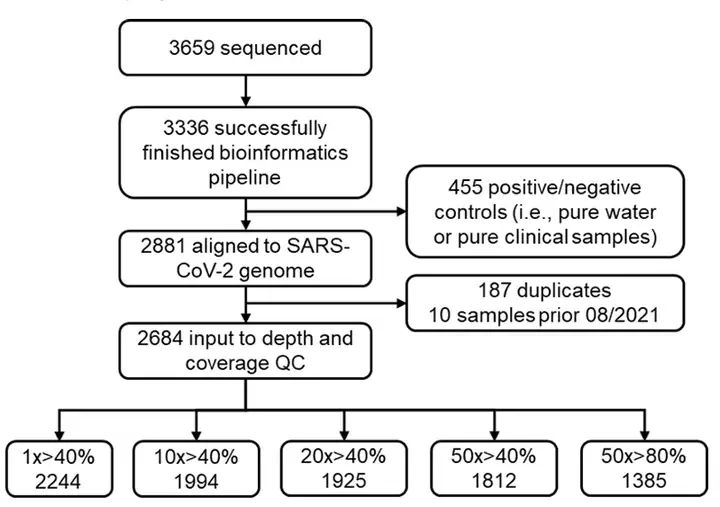
final,L’étude a utilisé 1 385 échantillons de haute qualité.Couverture de 59 422 sites de mutation des variants du SRAS-CoV-2 pour une analyse ultérieure.
Pour aider à vérifier l'efficacité de la méthode ICA-Var, l'équipe de recherche a utilisé des données cliniques comme base de contrôle et de référence et a analysé 8 810 données de séquence clinique à couverture élevée du SARS-CoV-2 du Nevada téléchargées à partir de la base de données GISAID, couvrant la période de septembre 2021 à novembre 2023.
Avec l'ICA comme noyau, une méthode de régression double est introduite pour créer un nouvel outil de détection du COVID-19
Le processus principal de l'ICA-Var estIl traite les fréquences de mutation dans les échantillons d'eaux usées grâce à une analyse des composants indépendants et extrait des modèles de mutation de covariation indépendants.Ces modèles sont ensuite associés aux échantillons d’origine par le biais d’une double régression pour suivre les variantes du virus, comme le montre la figure ci-dessous :
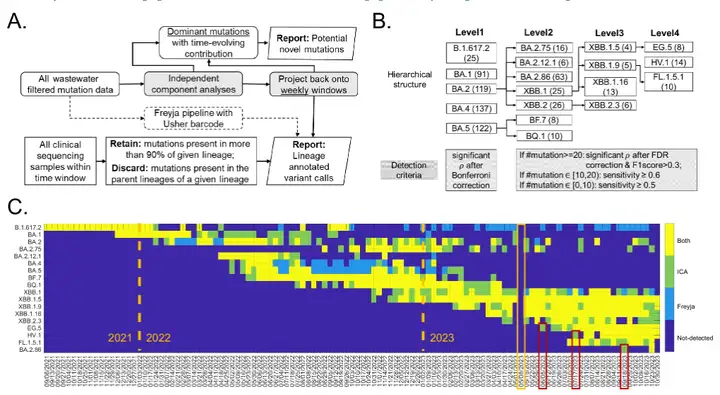
* A dans la figure est le processus d’analyse des composants indépendants.Les deux matrices sont : la détection hebdomadaire de la lignée du SARS-CoV-2 (rangée du bas) et les nouvelles mutations potentielles (rangée du haut).
* La figure B montre la structure hiérarchique de 18 variantes préoccupantes.Les principaux sites de mutation de chaque variante (c'est-à-dire les sites définissant la lignée) ont été prélevés http://covspectrum.org Données cliniques résumées, avec le nombre de mutations majeures entre parenthèses et les cases ombrées indiquant les critères à tester dans le flux de travail proposé.
*La figure C montre la comparaison entre la méthode ICA-Var et l’outil de pointe Freyja.Pour les variants nouvellement apparus EG.5, HV.1 et BA.2.86, la case rouge indique une heure de détection ICA-Var antérieure ; la case jaune indique une semaine au cours de laquelle l'échantillonnage des eaux usées n'a pas été effectué en raison de problèmes techniques.
Plus précisément, étant donné que le signal du génome du SRAS-CoV-2 dans les échantillons d’eaux usées est le résultat d’un mélange de plusieurs variantes et est perturbé par la dégradation de l’échantillon, les erreurs de séquençage, etc., les méthodes traditionnelles sont difficiles à analyser directement les caractéristiques d’une seule variante.L'idée principale d'ICA-Var est d'utiliser l'analyse des composants indépendants——Cette technologie de séparation de sources aveugle suppose que le signal de mutation mixte est une combinaison linéaire de plusieurs « sources indépendantes » et utilise une modélisation mathématique pour séparer ces modèles indépendants des données mixtes.
L’équipe de recherche a d’abord prétraité les données.En contrôlant la qualité des données de séquençage du génome du SARS-CoV-2 issues d'échantillons d'eaux usées, en filtrant les lectures de mauvaise qualité et les mutations bruyantes, une matrice de fréquence de mutation a été construite. Les lignes représentent les échantillons, les colonnes les sites de mutation et les valeurs la fréquence de chaque site. Une analyse en composantes indépendantes a ensuite été réalisée sur cette matrice, décomposant le signal mixte en composantes indépendantes. Chaque composante représente un ensemble de « schémas de mutation de covariation », ou combinaisons de mutations caractéristiques d'un variant particulier, qui apparaissent ou disparaissent de manière synchrone d'un échantillon à l'autre au fil du temps.
ici,L’étude a utilisé le critère de longueur de description minimale (MDL) pour déterminer le nombre de composants indépendants et a effectué une décomposition des composants indépendants à l’aide de l’algorithme fastICA.Pour garantir la fiabilité des résultats, ils ont répété l'analyse ICA 50 fois avec différentes valeurs initiales, ont regroupé et visualisé les composants obtenus à chaque exécution à l'aide du logiciel ICASSO, et ont finalement conservé uniquement les estimations fiables correspondant aux clusters serrés comme matrice source.
Ensuite, afin de déterminer davantage la situation des variantes hebdomadaires,L’équipe de recherche a utilisé la méthode de double régression pour reprojeter la matrice source obtenue à partir de l’analyse des composants indépendants dans l’échantillon d’origine.Calculez la « contribution » de chaque composant indépendant dans chaque échantillon, c'est-à-dire l'abondance relative de la variante dans l'échantillon, afin de quantifier les changements dynamiques des différentes variantes dans le temps et l'espace, tels que le moment d'apparition, les tendances épidémiques et les différences de distribution urbaine-rurale.
L'équipe de recherche a utilisé la matrice source de l'échantillon complet comme ensemble de régresseurs sources dans un modèle linéaire général (MLG) afin de déterminer les schémas de décomposition du signal pour chaque échantillon hebdomadaire en relation avec la matrice source de l'échantillon complet. Ils ont ensuite utilisé les schémas de décomposition du signal pour chaque échantillon hebdomadaire comme régresseurs dans un second MLG afin de déterminer la matrice source spécifique à la semaine, toujours en relation avec la matrice source de l'échantillon complet. Ce processus a généré des paires d'estimations constituant l'espace dual et, ensemble, a fourni la meilleure approximation de la matrice source de l'analyse en composantes indépendantes de l'échantillon complet d'origine pour chaque échantillon hebdomadaire.
enfin,L’équipe de recherche a comparé les composants indépendants isolés avec des variantes connues dans les données de séquençage clinique et les a annotés.Cela peut déterminer avec succès la souche variante correspondante ou éliminer les modèles de mutation de covariance non appariés pour avertir de la possibilité de nouvelles souches variantes.
La méthode ICA-Var surmonte les inconvénients des méthodes traditionnelles qui s'appuient sur des « codes-barres de variantes de référence prédéfinis ».En capturant les modèles de covariation des mutations, il est possible d’identifier de nouvelles variantes plus tôt et plus précisément que les méthodes traditionnelles.Associée à une analyse de régression double, cette méthode révèle également des différences de transmission urbaine et rurale ainsi que l'évolution temporelle des sites de mutation. En résumé, ICA-Var offre un outil plus sensible, plus complet et plus rentable pour la détection de la COVID-19.
L'efficacité de détection dépasse celle de l'outil de référence actuel Freyja et a le potentiel de prédire de nouvelles variantes
Pour valider et évaluer les performances d'ICA-Var, l'équipe de recherche l'a comparé à l'outil de référence actuel, Freyja, qui permet d'estimer l'abondance relative des lignées de SARS-CoV-2 dans les eaux usées. Cet outil utilise une bibliothèque de « codes-barres » composée de mutations définissant les lignées afin d'identifier de manière unique toutes les lignées de SARS-CoV-2 connues, et utilise une méthode de régression pondérée en profondeur et à écart absolu minimal pour calculer l'abondance des lignées.Des expériences ont confirmé que la méthode d’analyse multivariée ICA-Var présente des avantages plus significatifs.
Comme le montre la figure ci-dessous, la section sur la méthode et l'architecture du modèle explique brièvement comment ICA-Var peut détecter les nouvelles variantes EG.5, HV.1 et BA.2.86 plus tôt, et le contenu principal sera développé dans cette section.
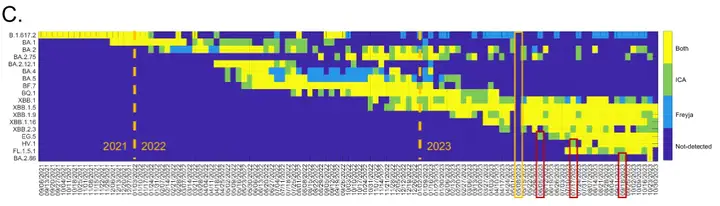
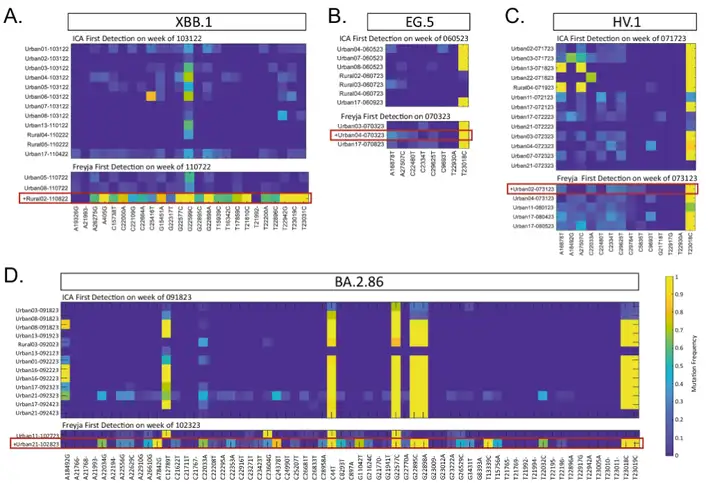
Plus précisément, en 2022,Il a été démontré que l'ICA-Var détecte des variantes telles que BA.2, BA.4, BA.5, BF.7, BQ.1, XBB.1 et XBB.1.5 une ou plusieurs semaines plus tôt que Freyja.Lors de la détection d'EG.5, ICA-Var a détecté ce variant la semaine du 5 juin, mais Freyja n'a identifié le signal d'EG.5 que le 3 juillet, lorsque l'abondance des échantillons d'eaux usées a atteint 23,08%, et que 5 des 8 sites de mutation dominants d'EG.5 étaient déjà présents. De même, pour des variants tels que XBB.1, HV.1 et BA.2.86,ICA-Var a également été détecté plusieurs semaines plus tôt que Freyja.
Cela est dû au fait que l’ICA-Var intègre des informations provenant de plusieurs échantillons sur « une prévalence fiable mais faible des sites de mutation dominants »., améliorant la puissance statistique et permettant une détection plus précoce. Cela signifie qu'il ne repose pas sur une forte proportion de mutations dominantes dans un seul échantillon ; il peut améliorer la détection simplement en agrégeant les signaux faibles de plusieurs échantillons. En revanche, Freyja nécessite qu'au moins un échantillon individuel montre clairement un site de mutation dominante pour une détection complète. Cela signifie également qu'il repose davantage sur un signal de mutation suffisamment fort dans un seul échantillon et est moins sensible aux signaux faibles ou dispersés.
L'expérience a également examiné les tendances dynamiques des variants dans les échantillons urbains et ruraux. Début 2022, l'équipe de recherche a séquencé et analysé des échantillons d'eaux usées provenant de zones rurales du sud du Nevada et a procédé à une comparaison épidémiologique complète entre zones urbaines et rurales, en analysant séparément les échantillons urbains et ruraux chaque semaine.
Les résultats ont montré que parmi les 18 variants préoccupants, ICA-Var et Freyja ont d'abord détecté 16 variants du SARS-CoV-2 dans des échantillons d'eaux usées urbaines avant d'être détectés dans des échantillons ruraux, ce qui indique que les variants du virus apparaissent généralement d'abord dans les villes, puis se propagent aux zones rurales. Comme le montre la figure ci-dessous :
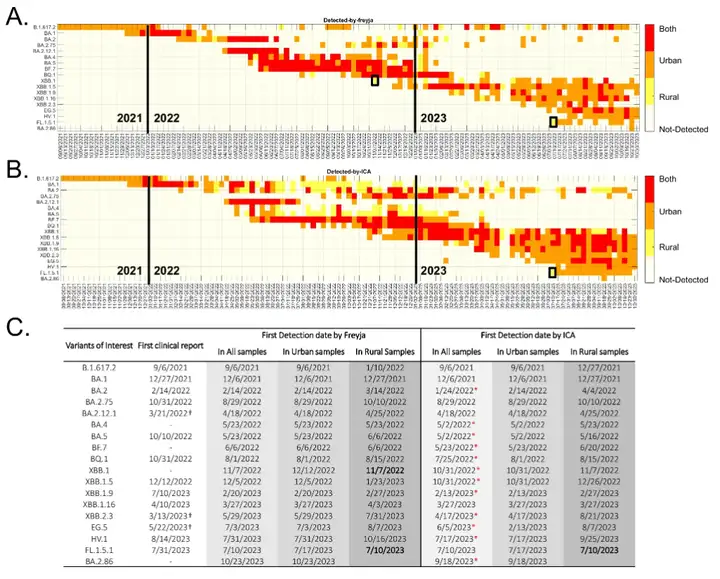
L'exception est que Freyja a initialement détecté XBB.1 dans des échantillons d'eaux usées rurales, tandis qu'ICA-Var a découvert la variante dans des échantillons d'eaux usées urbaines une semaine plus tôt ; les deux outils ont trouvé FL.1.5.1 dans des échantillons d'eaux usées rurales, tandis que la fréquence et la prévalence de l'allèle alternatif de la mutation dominante de cette variante étaient beaucoup plus faibles dans les échantillons d'eaux usées urbaines au cours de la même période.
L'étude a également révélé les tendances évolutives temporelles des sites de mutation. L'équipe de recherche a comparé 177 sites de mutation ayant contribué significativement à l'évolution temporelle entre août 2021 et novembre 2023 aux sites de mutation dominants des variants B.1.617.2, BA.1 et XBB.1, comme le montre la figure ci-dessous :
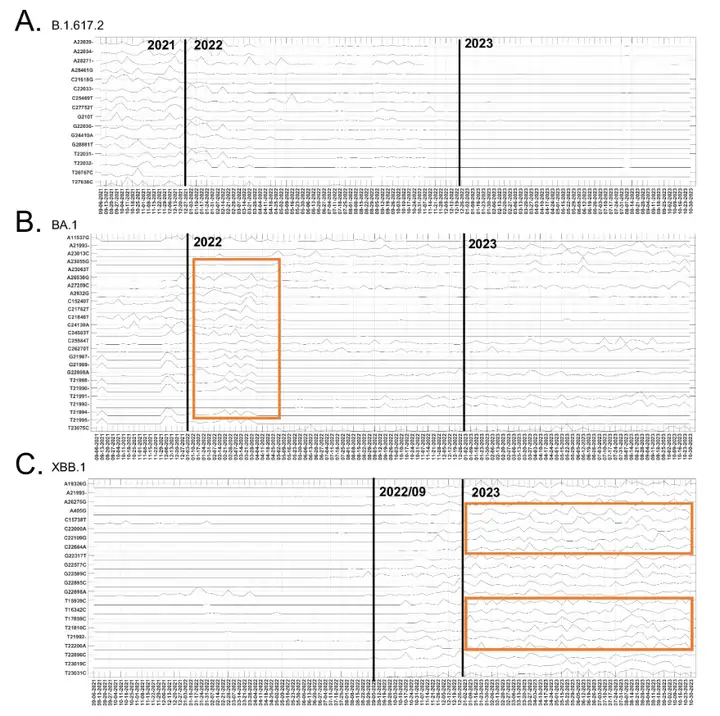
Français Sur les 25 principaux sites de mutation du variant Delta (B.1.617.2), 16 ont montré des fluctuations significatives de contribution à la fin de 2021, suivies d'un déclin progressif en 2022. La contribution des mutations apparentées dans le sous-type BA.1 d'Omicron a augmenté de manière significative à la fin de 2021 et a culminé début 2022. La contribution de certains sites de mutation BA.1 a continué de fluctuer en 2023 et a été trouvée dans d'autres sous-lignées d'Omicron, telles que XBB.1. Sur les 25 principales mutations du variant XBB.1, 22 ont montré des contributions dynamiques temporelles significatives, avec un impact significatif après septembre 2022. Plusieurs sites de mutation ont présenté des schémas de fluctuation similaires, indiquant une covariance, reflétant les caractéristiques de recombinaison de XBB.1.
Ces analyses ont démontré que les contributions évolutives temporelles des sites de mutation identifiés par ICA-Var étaient cohérentes avec les résultats cliniques des variantes Delta, Omicron et XBB.1, illustrant davantage la fiabilité des résultats d'ICA-Var et démontrant son potentiel à identifier de nouveaux modèles mutationnels qui peuvent conduire à l'émergence de nouvelles variantes.
L'expérience a procédé à une vérification détaillée de ce phénomène. L'équipe de recherche a examiné 113 nouveaux sites de mutation potentiels en les comparant aux sites de mutation dominants de 15 variants majeurs. Elle a ensuite utilisé un algorithme de classification hiérarchique pour classer ces sites de mutation en six groupes caractéristiques. Comme le montre la figure ci-dessous :

Parmi ces groupes caractéristiques, les sites de mutation de quatre d'entre eux (groupes 2 à 5) chevauchent ceux des variants apparus fin 2023. Les groupes 1 et 6 ne présentent aucune mutation chevauchante avec des sites de mutation connus. Parmi eux, les sites de mutation du groupe 1 ont montré un schéma de covariance évident après août 2023. Les données de séquençage clinique du GISAID ont montré que huit des sites de mutation ont été vérifiés et présentaient une faible fréquence de signalement dans les échantillons cliniques. Par conséquent,Ces mutations peuvent conduire à l’émergence de nouveaux variants du coronavirus, qui doivent être davantage vérifiés par des tests cliniques.Une surveillance étroite est nécessaire.
Grâce à l'apprentissage automatique, la surveillance des eaux usées continue d'évoluer pour favoriser une prévention et un contrôle des virus de haute qualité
Comme mentionné au début, l'analyse des eaux usées n'est pas une méthode nouvelle. Dès les années 1940, les virologues environnementaux ont reconnu l'intérêt d'obtenir le poliovirus par des expériences de culture cellulaire dans les eaux usées. Depuis, l'analyse des eaux usées n'a cessé d'être améliorée et est devenue un outil efficace d'alerte précoce en cas d'épidémie.Depuis le début de la pandémie de COVID-19, WBE a une fois de plus joué un rôle positif dans la prévention et le contrôle de l’épidémie.
Par exemple, fin 2023, des rapports ont indiqué qu'une équipe de recherche suédoise avait détecté avec succès l'émergence du nouveau variant BA.2.86 du SARS-CoV-2 en intégrant des tests génomiques sur les eaux usées et les cas de COVID-19. Par ailleurs, afin d'utiliser plus efficacement et activement l'analyse des eaux usées pour la détection de nouveaux variants du coronavirus, de nombreux laboratoires ont également développé ou amélioré des modèles connexes afin de fournir des outils plus rentables pour l'analyse des eaux usées.
Par exemple, des chercheurs de l'Université Tsinghua, de l'Université des sciences et technologies du Hebei et du Centre de surveillance écologique et environnementale de Tianjin ont publié conjointement une étude intitulée « Validation des méthodes d'enrichissement et de détection de l'ARN du SRAS-CoV-2 dans les eaux usées ». L'étude a comparé deux techniques d'enrichissement, l'ultrafiltration et la séparation par résine d'affinité covalente, à deux méthodes de détection, la PCR quantitative par transcription inverse (RT-qPCR) et la PCR numérique par transcription inverse (RT-dPCR), afin d'évaluer leurs performances dans la surveillance des virus dans les eaux usées.
enfin,L’étude a indiqué que la méthode de PCR numérique par transcription inverse (RT-dPCR) est un meilleur choix pour détecter de faibles concentrations d’ARN du SRAS-CoV-2 dans les eaux usées.Le taux de détection est plus élevé et il présente une meilleure tolérance aux inhibiteurs de PCR.
* Adresse du papier :
https://link.springer.com/article/10.1007/s10311-025-01843-6
Par ailleurs, l'équipe dirigée par le professeur Xingfang Li, du Département de pathologie et de médecine de laboratoire de l'Université de l'Alberta (Canada), a publié une étude intitulée « Quantification et différenciation des variants du SRAS-CoV-2 dans les eaux usées à des fins de surveillance ». S'appuyant sur les méthodes de détection par RT-qPCR multiplex Gamma (ABG) et Delta précédemment développées pour les échantillons cliniques, ils ont ciblé le sous-variant Omicron et exploité ses mutations spécifiques.Un test Omicron triplex RT-qPCR a été développé, capable de distinguer cinq sous-lignées majeures de variantes d'Omicron.Il s’agit de la première étude à utiliser un test triplex RT-qPCR à tube unique pour détecter et identifier toutes les sous-variantes d’Omicron dans des échantillons d’eaux usées sur une période d’un an.
* Adresse du papier :
https://pubs.acs.org/doi/10.1021/envhealth.3c00089
En résumé, le monde est aujourd'hui confronté à de graves défis en matière de santé et de sécurité publiques, et la surveillance des eaux usées, outil très efficace de surveillance de la population, joue un rôle irremplaçable. Grâce aux progrès technologiques constants, la surveillance des eaux usées continuera d'évoluer, allant de la détection précoce ciblée basée sur des profils de mutation connus aux avancées en matière de séquençage du génome entier et d'identification d'agents pathogènes inconnus. Grâce à une sensibilité et une couverture en constante augmentation, la surveillance des eaux usées fournira des données plus précises et essentielles pour l'alerte épidémique, le traçage et l'élaboration des politiques, devenant ainsi un complément essentiel à la ligne de défense en matière de santé et de sécurité publiques.
Références :
1.https://www.nature.com/articles/s41467-025-61280-5
2.https://mp.weixin.qq.com/s/ZzzZt-uNNc5DsD-ib3Ww8g








