Command Palette
Search for a command to run...
Sélectionné Pour NeurIPS 2024 ! L'Université De Westlake a Proposé Le Modèle Universel De Pliage Inverse Moléculaire UniIF, Qui Complète Encore AlphaFold 3.

Le repliement inverse moléculaire joue un rôle clé dans la conception de médicaments et de matériaux, permettant aux scientifiques de synthétiser de nouvelles molécules avec les structures souhaitées. Les études passées se sont principalement concentrées sur le repliement inverse des macromolécules ou des petites molécules, mais rarement sur le repliement inverse des molécules générales.
La construction d’un modèle général unifié comporte trois défis majeurs :1 Différences entre les unités : les grosses molécules utilisent généralement des microstructures prédéfinies comme unités de base, telles que les acides aminés pour les protéines et les nucléotides pour l'ARN ; les petites molécules utilisent des atomes comme unités de base ; 2. Extraction de caractéristiques géométriques : Différentes études utilisent diverses stratégies pour l’extraction de caractéristiques géométriques, telles que la distance, l’angle et le produit tensoriel, et manquent d’une méthode de caractérisation unifiée ; ③ Échelle du système : les petites molécules permettent aux mécanismes d’attention globale d’apprendre les dépendances à long terme, mais cela ne fonctionne souvent pas sur les grosses molécules.
Pour relever les défis ci-dessus et compléter davantage les progrès réalisés par RoseTTAFold All-Atom et AlphaFold 3 dans la prédiction de la structure moléculaire,Une équipe du Future Industry Research Center de l'Université Westlake a proposé un modèle unifié, UniIF, pour le repliement inverse de toutes les molécules.Les chercheurs ont mené des expériences complètes sur plusieurs tâches, notamment la conception de protéines, la conception d'ARN et la conception de matériaux, pour démontrer l'efficacité d'UniIF. Les résultats montrent qu’UniIF atteint des performances de pointe sur toutes les tâches.
La recherche connexe, intitulée « UniIF : Unified Molecule Inverse Folding », a été sélectionnée pour la conférence NeurIPS 2024.
Points saillants de la recherche :
* Le modèle unifié proposé UniIF fournit une solution polyvalente et efficace pour le repliement inverse moléculaire général
* Le modèle est unifié à deux niveaux : au niveau des données, un formulaire de données de graphe de blocs unifié pour toutes les molécules est proposé, y compris la construction du système de coordonnées local et l'initialisation des caractéristiques géométriques ; au niveau du modèle, un réseau d'attention par blocs géométriques est introduit pour capturer les interactions tridimensionnelles de toutes les molécules
* Les chercheurs ont démontré que la méthode proposée surpasse les méthodes de pointe dans trois tâches principales : la conception des protéines, la conception de l’ARN et la conception des matériaux. Cette réalisation pourrait avoir un impact positif sur les communautés de l’apprentissage automatique, de la découverte de médicaments et de la science des matériaux.
Adresse du document :
https://arxiv.org/abs/2405.18968
Suivez le compte officiel et répondez « Molecular Reverse Folding » pour obtenir le PDF complet
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensemble de données : sélectionnez l’ensemble de données correspondant pour trois expériences de tâches
Dans la tâche de conception de protéines,Les chercheurs ont évalué UniIF sur l’ensemble de données CATH4.3. L'ensemble de données est divisé selon le code de classification topologique CATH, ce qui donne 16 631 échantillons d'entraînement, 1 516 échantillons de validation et 1 864 échantillons de test.
Pour évaluer la capacité de généralisation, les chercheurs ont adopté une stratégie de partitionnement temporel, considérant que certaines lignes de base utilisent des modèles ESM2 pré-entraînés, ce qui présente un risque de fuite de données. L'évaluation partitionnée dans le temps attribue les données antérieures à une certaine date à l'ensemble d'apprentissage et les données postérieures à cette date à l'ensemble de test. Pour l'évaluation temporelle des structures, l'ensemble de données CASP15 est utilisé, qui contient de nouvelles structures cristallines non observées pendant la formation ; pour l'évaluation temporelle des séquences, l'ensemble de données NovelPro est utilisé, qui contient 76 séquences protéiques publiées dans les 30 jours précédant le 23 novembre 2023, et les structures sont prédites par AlphaFold 2.
Dans les tâches de conception d'ARN,Les chercheurs ont mené des expériences sur l'ARN sur un ensemble de données collectées par RDesign, qui contient 2 218 structures tertiaires d'ARN, qui sont divisées en un ensemble d'entraînement (1 774 structures), un ensemble de test (223 structures) et un ensemble de validation (221 structures) en fonction de leur similitude structurelle. En raison du petit nombre d’échantillons de données, les chercheurs ont rapporté le taux de récupération médian et son écart type de 3 exécutions indépendantes.
Dans la tâche de conception matérielle,Les chercheurs ont évalué UniIF sur l’ensemble de données CHILI-3K, qui se compose d’images de nanomatériaux dérivés d’oxydes métalliques simples. L'ensemble de données comprend 53 éléments métalliques et un élément non métallique (l'oxygène), avec un total de 3 180 graphiques, 6 959 085 nœuds et 49 624 440 arêtes.
Architecture du modèle : UniIF, un modèle unifié pour le repliement inverse moléculaire général
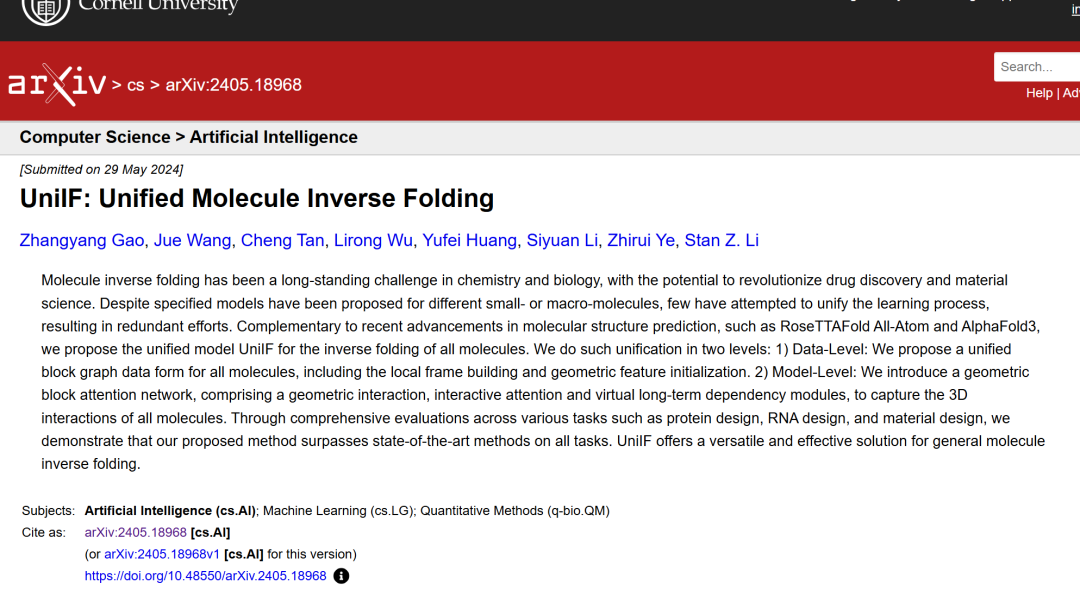
Comme le montre la figure ci-dessous, les chercheurs ont proposé un modèle unifié pour le repliement inverse moléculaire général.
1 Le modèle convertit tous les types de molécules en graphiques de blocs - pour les macromolécules, un cadre prédéfini basé sur les acides aminés et les nucléotides est utilisé ; pour les petites molécules, un cadre local pour chaque bloc est appris via une couche de GNN ;
2. Utilisez le Geometric Featurizer pour initialiser les entités de nœud géométrique et les entités de bord ;
③ Une couche d'attention Block Graph est proposée, sur la base de laquelle un réseau neuronal Block Graph est construit pour apprendre à exprimer des représentations de blocs riches ;
④ Enfin, nous démontrons qu’UniIF peut obtenir des résultats compétitifs dans une variété de tâches, notamment la conception de protéines, la conception d’ARN et la conception de matériaux.
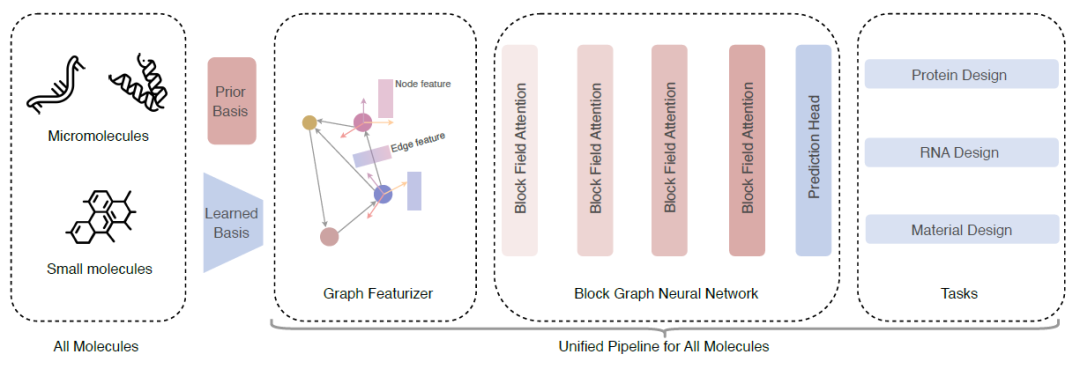
Schéma de principe :La première étape de l’architecture du modèle consiste à introduire des diagrammes en blocs pour représenter tous les types de molécules. La clé est de convertir des ensembles irréguliers d’atomes (de tailles variables) en représentations de blocs régulières (de taille fixe). Les chercheurs ont introduit une représentation par blocs basée sur des cadres pour unifier la modélisation de toutes les molécules, où un bloc contient un cadre équivariant et des vecteurs propres invariants, et un cadre local contient une matrice d'axes et un vecteur de déplacement. Pour les grosses molécules, la matrice d'axes est prédéfinie en fonction des acides aminés et des nucléotides ; pour les petites molécules, puisqu'il n'existe pas de modèle structurel commun a priori pour les petites molécules, la matrice des axes doit être apprise. Étant donné une molécule composée de n blocs, les chercheurs ont utilisé l’algorithme kNN pour construire un graphe de blocs.
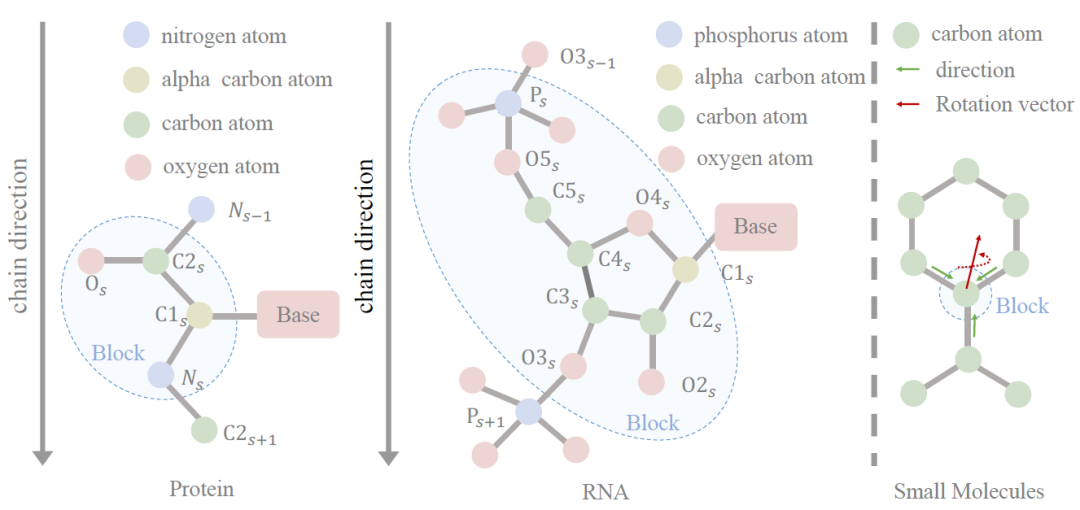
Extraction de caractéristiques de graphe en blocs :Pour les petites molécules, les cadres locaux prédéfinis ne sont pas disponibles, les chercheurs doivent donc apprendre un cadre local pour chaque atome - c'est-à-dire, étant donné une molécule, ils utilisent une couche de GNN pour initialiser la représentation atomique, puis utilisent un extracteur de caractéristiques géométriques pour initialiser les caractéristiques des nœuds géométriques et les caractéristiques des bords.
Module d'attention du graphique en blocs :Les chercheurs ont introduit un réseau d’attention par blocs géométriques, comprenant des modules d’interaction géométrique, d’attention d’interaction et de dépendance virtuelle à long terme, pour capturer les interactions tridimensionnelles de toutes les molécules.
Résultats de recherche : UniIF surpasse les méthodes de pointe sur toutes les tâches
Les chercheurs ont démontré l'efficacité d'UniIF à travers de multiples tâches de pliage inverse et des études d'ablation, notamment :
* Conception de protéines (T1) : Conception de séquences protéiques capables de se replier dans des structures cibles
* Conception d'ARN (T2) : Concevoir des séquences d'ARN qui peuvent se replier dans des structures cibles
* Conception matérielle (T3) : Découverte de compositions stables à partir de structures matérielles connues
1 Conception de protéines (T1)
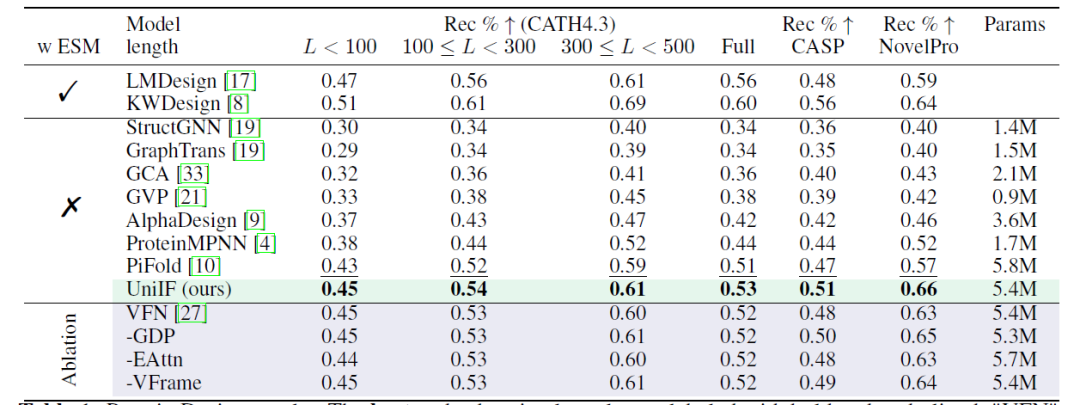
La conception de protéines vise à concevoir des séquences de protéines qui peuvent se replier dans une structure cible, et les chercheurs fournissent des résultats dans différents contextes (avec et sans ESM2) et plusieurs ensembles de données (CATH4.3, CASP, NovelPro). Comme le montre le tableau suivant : en utilisant un modèle de pliage inverse pur sans ESM2, UniIF obtient les meilleures performances sur tous les ensembles de données, démontrant ainsi son efficacité.
*LMDesign et KWDesign incluent ESM2 ; StructGNN, GraphTrans, GCA, GVP, AlphaDesign, ProteinMPNN et PiFold n'incluent pas ESM2
Sur CATH4.3, en raison du modèle de base solide, l'amélioration globale est limitée, mais l'évaluation partitionnée dans le temps met en évidence l'avantage d'UniIF dans la capacité de généralisation. UniIF surpasse le PiFold de base solide avec moins de paramètres d'apprentissage. Dans l'évaluation du partitionnement temporel, UniIF surpasse toutes les lignes de base, y compris l'approche basée sur ESM2, par une marge significative. Sur NovelPro contenant de nouvelles séquences, UniIF a surpassé LMDesign et KWDesign en utilisant ESM2 pour l'optimisation des séquences.Cela indique qu'UniIF possède une capacité de généralisation supérieure, ce qui est crucial pour les applications pratiques.
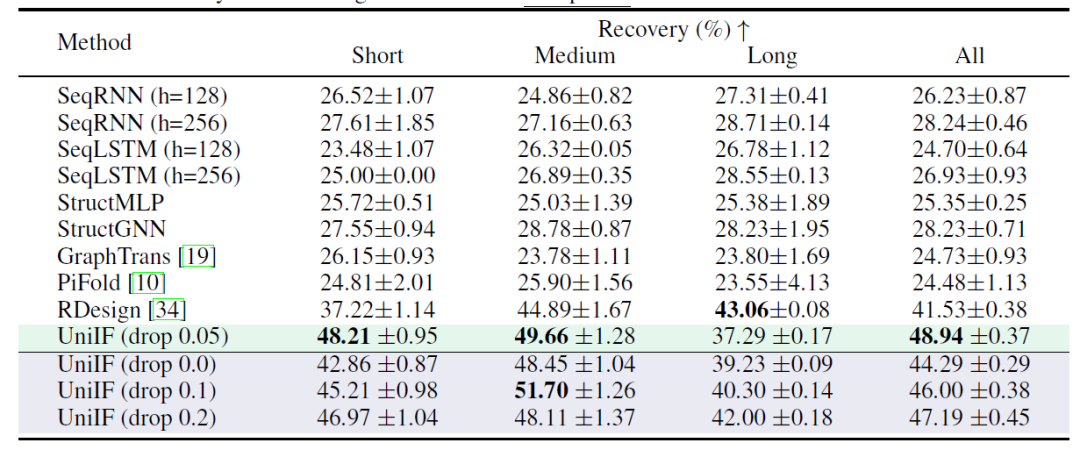
2 Conception d'ARN (T2)
L’objectif de la conception d’ARN est de concevoir des séquences d’ARN qui peuvent se replier dans une structure cible. Comme le montre le tableau ci-dessous, UniIF obtient les meilleures performances dans tous les cas, ce qui constitue une amélioration significative puisque les modèles de base auparavant solides tels que PiFold n'excellaient que dans la conception de protéines. Il est rapporté que,UniIF est le premier modèle à atteindre des performances de pointe dans les tâches de conception de protéines et d'ARN, démontrant ainsi sa polyvalence et son efficacité.
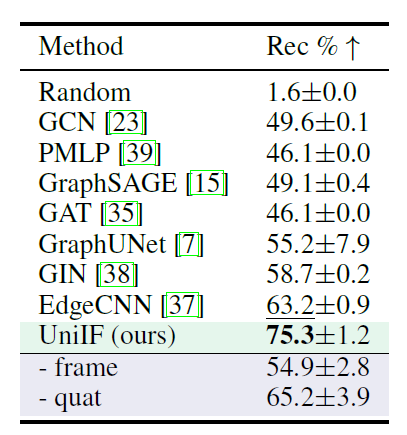
③ Conception matérielle (T3)
Trouver des combinaisons atomiques stables au sein de structures matérielles connues est crucial pour découvrir de nouveaux matériaux. Les chercheurs ont donc également évalué les performances de l'UniIF sur cette nouvelle tâche. Comme le montre le tableau suivant,UniIF surpasse considérablement tous les modèles de base.
④ Étude de cas
Dans la figure ci-dessous, les chercheurs montrent les séquences de protéines et d’ARN conçues. De plus, la séquence conçue a été repliée en structure à l'aide d'AlphaFold 3 - la vraie structure (gris), la structure PiFold (vert) et la structure UniIF (rose) ont été alignées et comparées. Les chercheurs ont observé queUniIF obtient des améliorations à la fois du taux de récupération et de l'écart quadratique moyen (RMSD), démontrant son efficacité dans la tâche de pliage inverse.

Le modèle UniIF complète encore AlphaFold 3
L’apprentissage moléculaire général a reçu une attention croissante ces dernières années, et RoseTTAFold All-Atom (RFAA) et AlphaFold 3 sont deux modèles représentatifs qui ont obtenu un succès remarquable dans cette direction.
Le 7 mars 2024, David Baker a publié un article de recherche intitulé « Modélisation et conception biomoléculaires généralisées avec RoseTTAFold All-Atom » dans Science. L'équipe a développé RoseTTAFold All-Atom (RFAA), qui combine des représentations basées sur des résidus d'acides aminés et de bases d'ADN avec des représentations atomiques de tous les autres groupes pour modéliser des assemblages comprenant des protéines, des acides nucléiques, de petites molécules, des métaux et des modifications covalentes d'une séquence et d'une structure chimique données.
Article original :
https://www.science.org/doi/10.1126/science.adl2528
Le 9 mai 2024, Demis Hassabis, John Jumpe et d'autres ont publié un article de recherche intitulé « Prédiction précise de la structure des interactions biomoléculaires avec AlphaFold 3 » dans Nature. L'étude a lancé AlphaFold 3, un nouveau modèle capable de prédire la structure des complexes contenant presque tous les types moléculaires de la Protein Data Bank, y compris la manière dont les ligands (petites molécules), les protéines et les acides nucléiques (ADN et ARN) se rassemblent et interagissent les uns avec les autres, ainsi que de prédire les effets structurels des modifications post-traductionnelles et des ions sur ces systèmes moléculaires, aidant ainsi les chercheurs à observer avec précision la structure des systèmes moléculaires biologiques au niveau atomique.
Article original :
https://www.nature.com/articles/s41586-024-07487-w
En examinant de plus près les deux modèles, RFAA utilise des diagrammes atome-liaison pour représenter les petites molécules et des diagrammes de charpente pour représenter les grandes molécules ; AlphaFold 3 utilise une représentation à deux couches, à savoir la représentation des atomes et la représentation des étiquettes, qui s'applique à toutes les molécules. Le concept de balise est équivalent au concept de bloc mentionné ci-dessus, représentant un groupe d'atomes, tels que des acides aminés ou des nucléotides.
GET et EPT sont deux modèles récemment proposés qui adoptent une représentation par blocs pour les petites et grandes molécules et introduisent de nouveaux cadres de transformateurs isotropes. Contrairement à RFAA qui spécifie les diagrammes atome-liaison pour les petites molécules, le modèle UniIF introduit dans cet article adopte un diagramme de blocs unifié pour tous les types de molécules, qui ne nécessite pas de diagrammes atome-liaison. De plus, le modèle introduit également une base vectorielle pour chaque bloc, différente d'AlphaFold 3, GET et EPT.
Le défi de construire un modèle moléculaire universel ayant été résolu dans une certaine mesure,Le modèle UniIF peut être considéré comme un complément supplémentaire aux progrès réalisés dans la prédiction de la structure moléculaire par des « prédécesseurs » tels que RoseTTAFold All-Atom et AlphaFold 3.À l’avenir, l’itération continue des grands modèles biologiques aidera les chercheurs à recomprendre le monde biologique et à repenser la découverte de médicaments, bénéficiant ainsi à toute l’humanité.
Références :
1.https://arxiv.org/abs/2405.18968
2.https://mp.weixin.qq.com/s/8OvxVlUuZZZ2gcepIl5UBw
3.https://www.jiqizhixin.com/articles/2024-03-08-6
4.https://m.thepaper.cn/newsDetail_forward_28984037









