Command Palette
Search for a command to run...
Materials AI Is Moving Towards an "explainable Era": A Japanese Team Cracks the Black Box of high-dimensional Spectroscopy, Pinpointing Key Features for Discovering New materials.

In recent years, the application of machine learning in the field of materials science has attracted much attention. Its application has gradually expanded from early structure-property scalar prediction (such as band gap energy, point defect formation energy, melting point, etc.) to more complex high-dimensional physical quantity modeling. One of the most challenging directions is the prediction and analysis of material spectra.
Spectral data, such as dielectric functions, spectra (absorption, reflection, and emission), and electronic and phonon densities of states, are crucial for understanding and designing materials. However, compared to scalar properties, high-dimensional spectral data is characterized by large output dimensions, complex structures, and strong physical constraints, making it difficult for traditional machine learning methods to simultaneously achieve accuracy and interpretability. While deep learning models have been able to predict spectra to some extent, the lack of interpretability remains a key bottleneck restricting their further application in materials design.
In this context,A research team from the Tokyo Institute of Science in Japan has proposed a method for interpreting deep learning models that can handle high-dimensional spectral data in materials science.Researchers constructed a dataset of first-principles calculations of the optical absorption spectra of 2,681 oxides, chalcogenides, and related compounds. Compared with standard density functional calculations, the calculated results showed significantly improved agreement with reported experimental spectra after corrections to the spectral onset energy and shape.
The researchers also used the dataset and the ALIGNN algorithm to develop a high-precision optical absorption spectrum prediction model.By combining feature extraction and cluster analysis, the key element types and their coordination environments that mainly determine the light absorption initiation energy and intensity were successfully extracted.
The related research findings, titled "Deep Learning–Based Extraction of Promising Material Groups and Common Features from High-Dimensional Data: A Case of Optical Spectra of Inorganic Crystals," were published in Advanced Intelligent Discovery.
Research highlights:
* This study proposes a method for material classification by feature extraction and cluster analysis of high-dimensional spectral data, thereby extracting potential material groups and their common features.
* The first-principles calculation dataset and machine learning model developed in this study are expected to play an important role in future materials discovery and materials informatics research.
* The method proposed in this study has broad applicability and can be used for the classification and interpretation of various spectral data. Its application is not limited to the optical absorption spectra of inorganic crystals.

Paper address:https://advanced.onlinelibrary.wiley.com/doi/10.1002/aidi.202600007
Datasets constructed using high-throughput first-principles calculations
Researchers first screened oxides, chalcogenides and related materials from the Materials Project database that met the following conditions: (1) The material contains at least one of the elements O, S, and Se, and its oxidation number is not necessarily −2; (2) The material does not contain the following elements: H, halogens, rare gases, Mn–Ni, Tc–Rh, Os–Ir, Po, lanthanides (except La and Ce) and actinides; (3) The material does not exhibit spin polarization; (4) Systems with space group P1 and/or more than 40 atoms in the original unit cell were excluded due to high computational cost or uncertainty in crystal structure.
The total number of materials used in the first-principles calculations was 9,808, and the calculation database was constructed according to the process shown in the figure below.
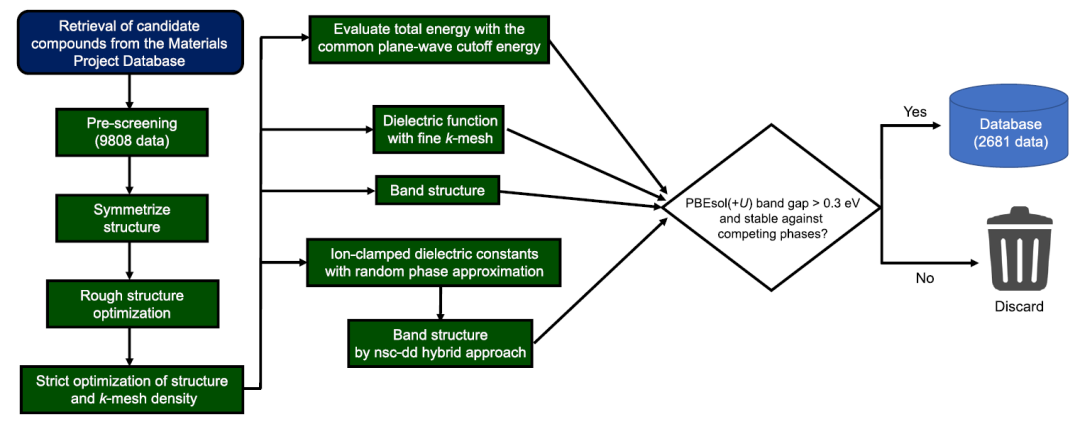
As shown in the figure, this calculation process is extremely complex. In order to achieve high-throughput computing while maintaining consistency and efficient use of computing resources,Researchers used their own developed program and relied on tools such as pymatgen, FireWorks, Custodian, atomate, and vise to automate the process.All first-principles calculations were performed using the VASP software package. This workflow uses PBEsol(+U) calculations to generate optical absorption spectra and compound formation energies, and uses nsc-dd mixed functionals and PBEsol(+U) calculations to obtain the band structure.
Regarding the machine learning dataset, the researchers removed: (1) materials that were unstable relative to the competing phase in the local database; and (2) materials with a PBEsol(+U) band gap of less than 0.3 eV. The final number of materials retained was 2681.
Constructing an ALIGNN model based on optical absorption spectra
Machine learning model construction and prediction accuracy
At the model level,This study uses ALIGNN (Atomistic Line Graph Neural Network) as the core prediction framework to model high-dimensional optical absorption spectra.Compared to traditional crystal graph convolutional networks (CGCNN), the core advantage of ALIGNN lies in the simultaneous introduction of dual representations of "atomic graph + bond line graph", thereby explicitly encoding three-body angle information and achieving a more refined expression of the local structural environment. The upper part of the figure below shows a schematic diagram of the ALIGNN architecture.
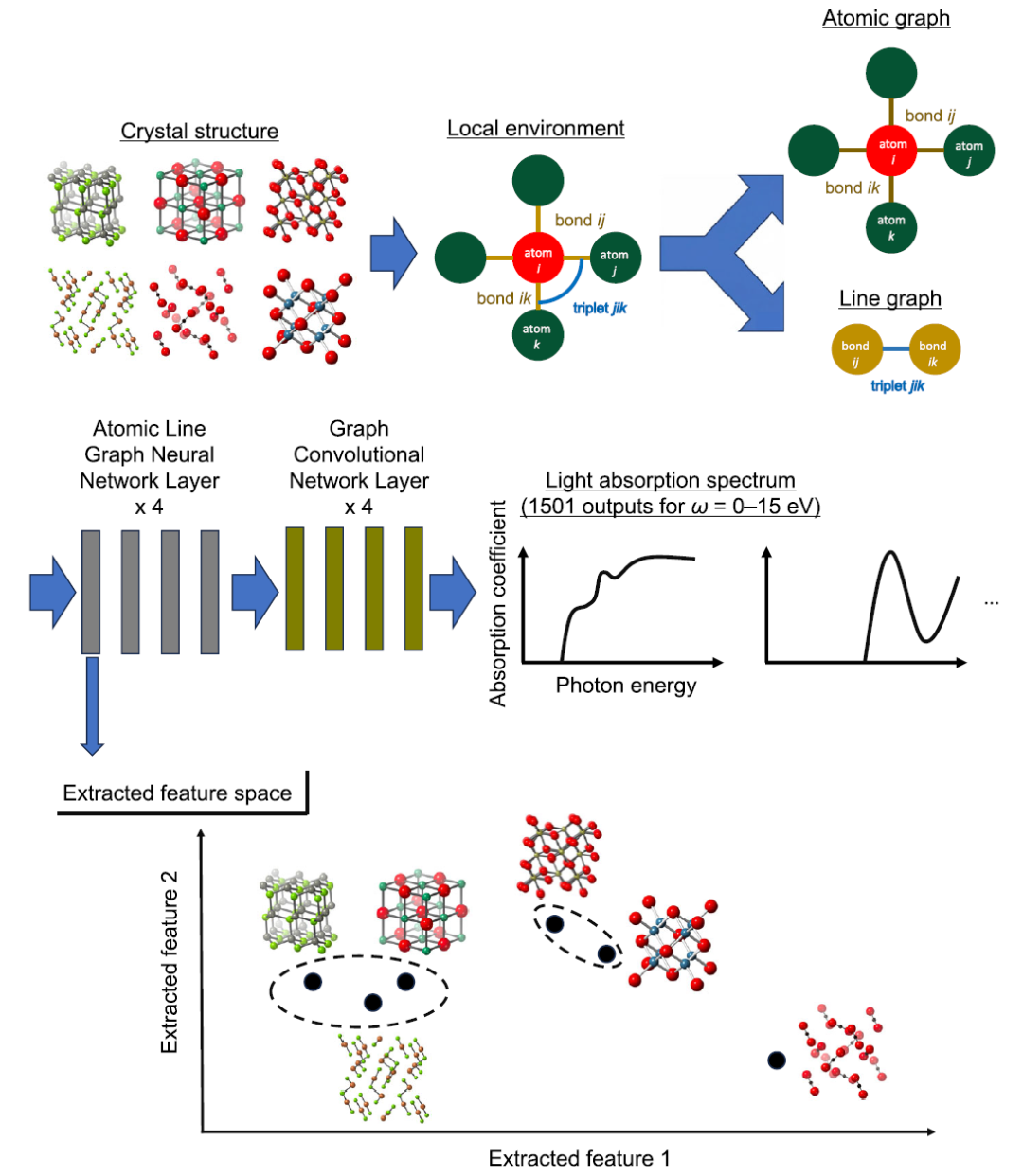
In this framework, atoms are nodes, inter-atomic bonds are edges, and the relationships between edges are further constructed as line graphs, thereby transforming bond angle information into learnable structural features.This design enables the model to not only capture the distance information between two bodies, but also to characterize the interaction between three bodies, thus more closely resembling the physical behavior of crystals.
Feature extraction and clustering
Researchers extracted features from the first layer of the optimized model's ALIGNN and averaged the feature vectors of all atomic sites for each material before performing hierarchical clustering analysis, as shown in the lower half of the figure above. The goal of this method is to classify materials into groups that exhibit similarity in both input features (such as elemental composition and atomic coordination features, including the number of adjacent atoms, interatomic distances, and bond angles) and output properties (optical absorption spectra).
The figure below shows the optical absorption spectra of the 96 groups obtained through hierarchical clustering. The spectral shapes within each cluster are indeed similar, confirming the effectiveness of the clustering method in this study.

Results: Interpretable extraction of material population structure and physical mechanisms was achieved.
To verify the ability of the new deep learning model to process high-dimensional spectral data in materials science, researchers conducted a series of experiments:
Predictive performance capability
In terms of prediction performance, the ALIGNN model demonstrated high accuracy across the test set, as shown in the figure below.The mean absolute error (MAE) of the material absorption spectrum prediction for approximately 75% is less than 0.14, indicating that the model can reproduce complex spectral shapes well.
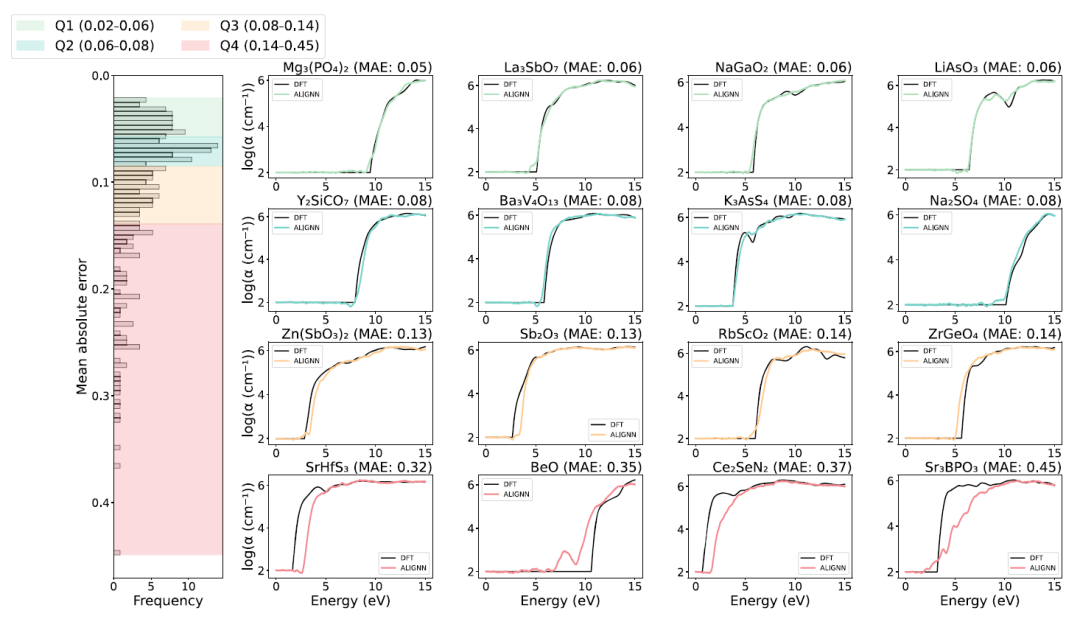
The right panel of the image above shows the prediction results for the four materials with the largest errors in each quartile. For the materials in the first three quartiles, the ALIGNN prediction results (colored curves) agree well with the first-principles reference calculation results (black curves); however, some compounds in the fourth quartile show significant deviations in the starting position of their optical absorption spectra. These outlier samples have poor prediction performance, mainly due to their unique electronic structures and the lack of similar structural materials in the training dataset.
The ability to capture the starting position of optical absorption spectra
Although MAE is a global metric covering the entire spectral range, researchers further examined whether the model could accurately reproduce the local spectral initiation energy. The figure below shows a parity plot: comparing the lowest photon energy corresponding to when log₁₀ α(ω) first exceeds 2.5 in first-principles calculations and ALIGNN predictions, where α represents the absorption coefficient.
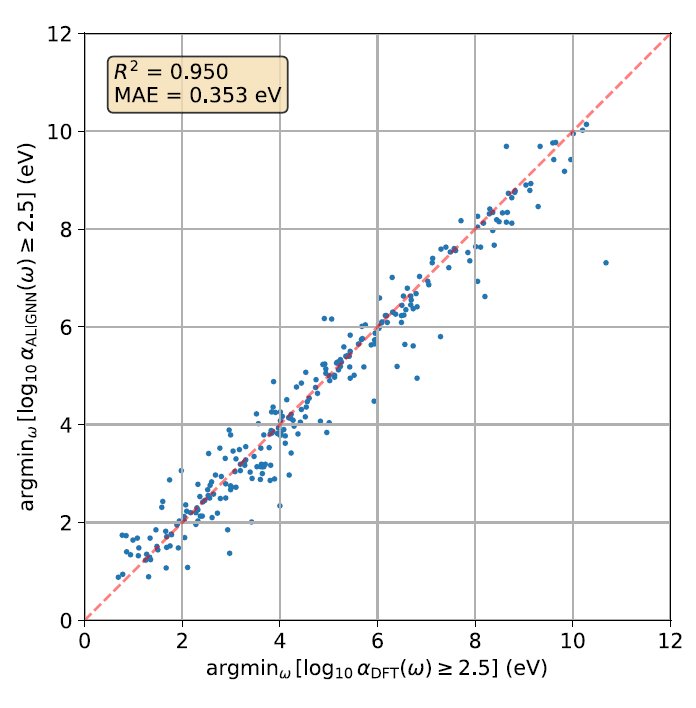
The results show that the predicted R² for the initial energy is 0.950 and the MAE is 0.353 eV, indicating that the ALIGNN model can accurately capture the starting position of the optical absorption spectrum.
Interpretability Analysis
In terms of interpretability analysis, researchers extracted feature representations from the first layer of ALIGNN and performed hierarchical clustering of the materials, resulting in 96 material groups. The results show that...Materials within the same cluster exhibit a high degree of consistency in spectral shape, particularly in the absorption initiation position and the steepness of the absorption edge, demonstrating significant commonality. This indicates that the model has learned spectral-related structural features in early layers.
Further case studies reveal clear physical differences between different material groups. For example, Group 74 typically exhibits broad band gaps and high absorption coefficients near the spectral inflection point. Figure a shows that all materials in this group contain either V or Cr, while other cations are primarily alkali metals. These materials mostly exist in the forms of VO₄³⁻, CrO₄²⁻, or Cr₂O₇²⁻, with the cations situated in a tetrahedral coordination environment.
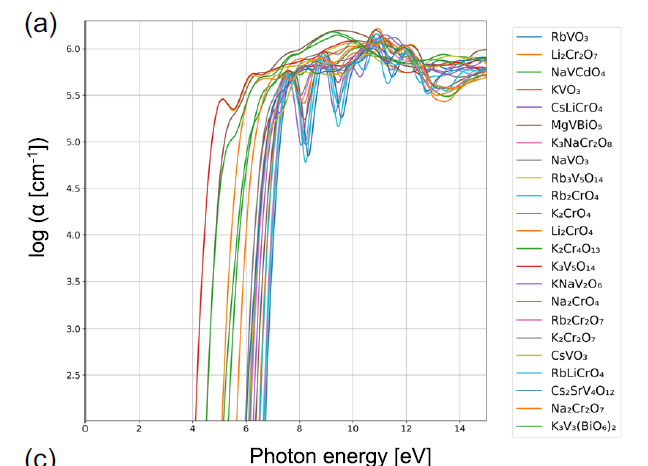
Optical absorption spectra of substances belonging to cluster 74, where α represents the absorption coefficient.
Researchers used CrystalFingerprintNN, implemented in Matminer, to calculate the tetrahedral coordination index of cation sites in each material within the cluster and analyzed the distribution of the maximum values for all cation sites. As shown in Figure b below, most materials do indeed possess tetrahedral coordination sites.
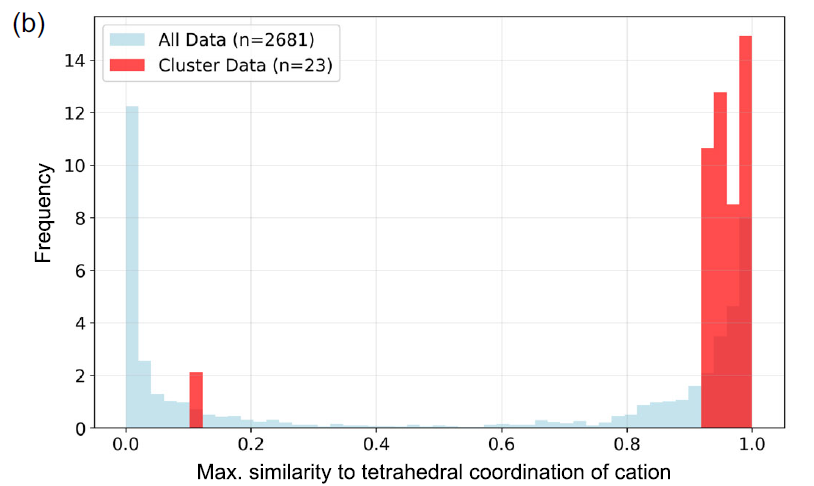
Tetrahedral coordination similarity distribution between the 74th cluster of materials (red) and the overall dataset (blue).
From the perspective of electronic density of states, a sharp peak caused by Vd or Cr-d states can be observed near the conduction band bottom (CBM). The high valence states of V⁵⁺ and Cr⁶⁺ provide a large number of unoccupied electronic states that can be used for optical transitions. Therefore, from the perspective of solid-state chemistry and physics, it is reasonable for these vanadates, chromates, and dichromates to have high optical absorption coefficients.
This process of inferring chemical mechanisms from model clustering results transforms machine learning outcomes from black-box predictions into a valuable source of knowledge for materials design. Furthermore, the study compared the results of direct clustering based on raw spectral data, finding that while it could identify similar spectra, it struggled to form clear chemical structure groups, resulting in significant mixing of material types. This further demonstrates the advantage of the ALIGNN feature space in achieving consistent structure-property representation.
Conclusion
The significance of this study lies not only in constructing a high-precision optical absorption spectroscopy prediction model, but more importantly, in proposing a methodological framework that combines "deep learning representation learning" with "materials physics interpretation." By combining the ALIGNN model with hierarchical clustering analysis, the study achieves the ability to extract common laws of materials from high-dimensional spectral data, enabling machine learning models not only to predict results but also to reveal the underlying structure and electronic origins of those results.
Ideally, the effects of electron-hole interactions, electron-phonon coupling, and point defects should be taken into account to reproduce the spectral characteristics of exciton effects, phonon-assisted electronic transitions, and defects, respectively. However, high-throughput first-principles spectral calculations including these effects are computationally too expensive, and therefore this was not achieved in this study. Even so, with the further integration of more precise many-body computation methods and machine learning models, this type of research is expected to play a more central role in materials discovery, propelling materials design from an experience-driven approach to a new stage that integrates data-driven and mechanism-driven approaches.