Command Palette
Search for a command to run...
The University of Toronto and Others Proposed dnaHNet, Which Improves Inference Speed by 3 Times and Reduces the Computational Cost of Genome Learning by Nearly 4 times.

The genome is the carrier of all the genetic information of an organism, determining cell function, individual development, and the direction of species evolution.The "DNA syntax" hidden within the sequence constitutes the underlying rules governing life and is one of the core problems that modern biology urgently needs to solve.Understanding this grammar is not only related to basic scientific knowledge, but also directly affects the development of key applications such as disease diagnosis, drug development, and synthetic biology.
In recent years, basic models pre-trained on large-scale sequence data have gradually become an important path to solving this problem. With the continuous improvement of computing power, data scale, and model parameters, these models have shown a performance growth trend similar to the "scaling law". Models represented by Nucleotide Transformer and Evo have expanded their parameter scale to the billions and have been trained on cross-species sequences, achieving significant progress in tasks such as variation effect prediction and regulatory element analysis.
However, DNA sequences are essentially continuous chains of nucleotides with indistinct boundaries, a fundamental difference from natural language. The two main modeling paradigms currently in use—Fixed word segmentation and single nucleotide level modeling each present a clear trade-off between expressive power and computational efficiency:The former may damage biological functional units, while the latter incurs high computational costs. Therefore, achieving a better balance between computational feasibility and biological fidelity has become a key challenge. Dynamic word segmentation, as a potential solution, still requires systematic exploration.
In this context,The dnaHNet model, jointly proposed by the University of Toronto, the Vector Institute for Artificial Intelligence in Canada, and the Arc Institute in the United States, among other institutions,This provides a new approach to overcoming the aforementioned bottlenecks. The related research results, titled "dnaHNet: A Scalable and Hierarchical Foundation Model for Genomic Sequence Learning," have been published as a preprint on arXiv.
Research highlights
* dnaHNet's computational efficiency surpasses StripedHyena2, and its inference speed is more than 3 times faster than Transformer.
* We propose optimal training strategies such as compression ratio scheduling and encoder-decoder balancing.
* Reached the forefront in zero-sample tasks such as variation effect prediction and gene necessity classification.
* It can learn context-dependent biological word segmentation and adapt to functional regions such as codons, promoters, and intergenic regions.

Paper address:
https://arxiv.org/abs/2602.10603
Follow our official WeChat account and reply "dnaHNet" in the background to get the full PDF.
Design of multi-level genomic datasets for model training and evaluation
To support model training and system evaluation, this study constructed a multi-layered data system.The pre-training data comes from a processed subset of the Genome Classification Database (GTDB).The process strictly followed the filtering, quality control, and redundancy removal procedures for the Evo model OpenGenome dataset. Screening criteria included key indicators such as assembly integrity, contamination level, and marker gene content; after screening, only one representative genome was retained for each species-level cluster.
The final dataset covers 85,205 prokaryotes and contains 17,648,721 sequences.The total number of nucleotides is approximately 144 billion. All sequences were extracted from the complete genome and divided into non-overlapping segments of up to 8,192 nucleotides.
In terms of evaluation, researchers constructed a test set from three complementary dimensions to comprehensively examine the model's capabilities. First,At the level of local coding fitnessTwelve nucleotide-level experimental datasets, totaling 21,250 data points, derived from E. coli K12 in MaveDB were used to evaluate the model's ability to characterize local coding grammar and protein fitness landscape.
Secondly,In terms of functional assessment at the whole genome scaleBased on the Essential Gene Database (DEG), binary essentiality tags were constructed for 62 bacterial species. Relevant sequences and annotations were obtained from NCBI, and a sequence identity greater than 99% with the DEG entry name was used as the essential gene labeling standard. This resulted in 185,226 data points, which were used to evaluate the model's ability to integrate long-range dependencies and genomic context.
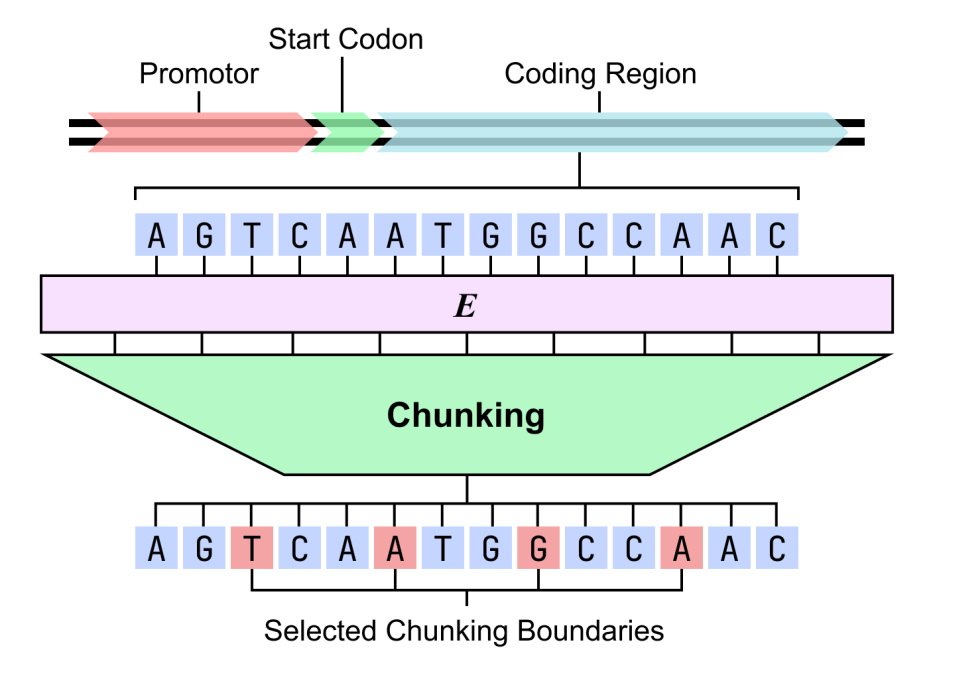
at last,In terms of structural interpretability,Taking the Bacillus subtilis genome as an example, the sequence is divided into different functional regions by combining its functional annotation. The structural modeling ability is verified by analyzing the alignment between the model segmentation results and the real biological structures.
dnaHNet model: Autoregressive frontier model without word segmentation
dnaHNet is a genome-based model that does not require an explicit segmenter.The key lies in the "dynamic segmentation" mechanism, which allows the model to learn the structural units in the sequence on its own.This design avoids the fragmentation of biological functional segments by fixed word segmentation and alleviates the computational overhead of nucleotide-by-nucleotide modeling, thus achieving a better balance between expressive power and computational efficiency.
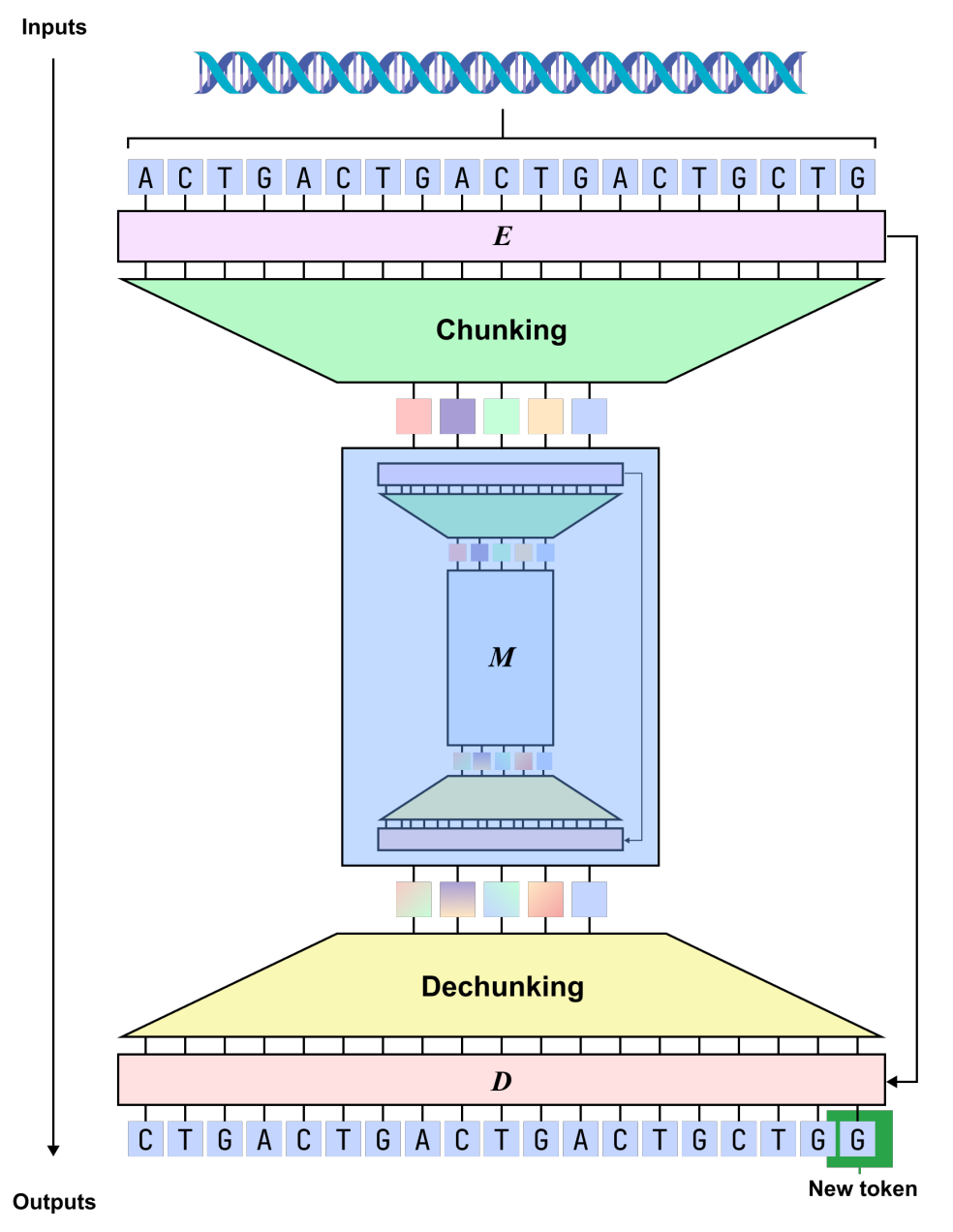
In terms of modeling form,dnaHNet unifies genome learning into an autoregressive sequence prediction task, which predicts the next nucleotide based on the existing context.The overall architecture adopts a hierarchical structure, with each layer consisting of an encoder, a backbone network, and a decoder: the encoder identifies locations in the sequence where information changes significantly (such as codon boundaries or regulatory regions) through a routing mechanism, and compresses the sequence into an implicit block representation accordingly; the backbone network adopts a hybrid structure combining Mamba and Transformer to model the compressed sequence, taking into account both long-range dependencies and key information interactions; the decoder then upsamples the representation back to nucleotide resolution and outputs the prediction results.
Building upon this foundation, dnaHNet underwent several key optimizations for genomic data. First, in terms of parameter allocation, approximately 301 TP3T of model capacity was allocated to the encoder and decoder to enhance the ability to characterize local structures.
Secondly,Incorporating a two-stage layered compression design:The first stage focuses on capturing short-scale patterns (such as codons), while the second stage models a longer range of functional structures, thus achieving a balance between compression efficiency and information fidelity. Furthermore, the training process incorporates autoregressive prediction loss and compression ratio constraints, enabling the model to effectively control computational costs while maintaining prediction accuracy.
During the inference phase, the model dynamically determines the block division method based on the boundary probability, so that the modeling granularity can adapt to the context and thus more closely resemble the real genome structure.
dnaHNet reduces computational cost by 3.89 times and outperforms other multi-task performers.
To systematically evaluate the performance of dnaHNet, this study compared it with two mainstream long-sequence genome models: StripedHyena2 and Transformer++.The experiments cover multiple aspects, including scaling properties, zero-sample variation effect prediction, gene necessity prediction, and biological structure modeling.
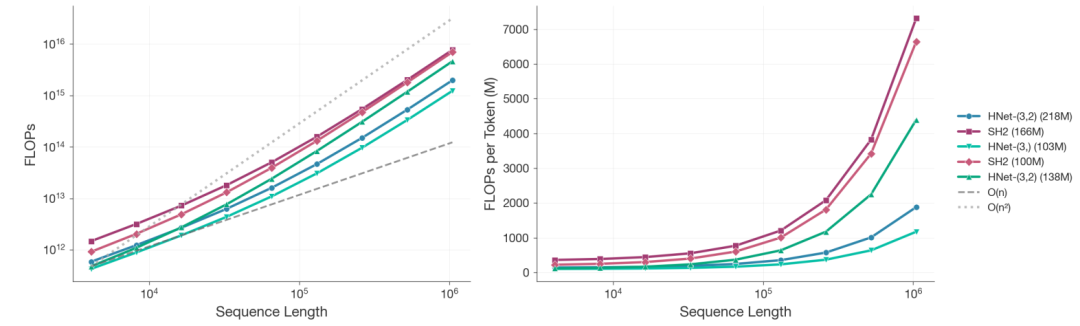
In scaling analysis,Researchers trained over 100 models of varying sizes under a fixed computing budget. When the sequence length reached 10⁶ nucleotides and the total computing power was 8 × 10¹⁹ FLOPs,The computational cost of dnaHNet with 218M parameters is reduced by approximately 3.89 times compared to StripedHyena2 with 166M parameters.The two-stage structure is even more efficient than the single-stage version.
The power-law fitting results based on perplexity show that,To achieve the same performance level, StripedHyena2 requires approximately 3.75 times the computing power of dnaHNet.Furthermore, dnaHNet's optimal data-parameter configuration deviates significantly from the traditional scaling law: under the same computing power, its training token count can reach 140B, while the comparison model only has 68B and has not yet converged.
In downstream tasks,dnaHNet consistently outperforms the comparison models in both zero-sample protein variation effect prediction (MaveDB) and gene necessity prediction (DEG).Furthermore, its advantages expand further with increased computing power. This indicates that its dynamic block-based mechanism and hierarchical architecture can more effectively integrate local coding syntax and global contextual information, thereby enhancing its ability to characterize biological functions.
Regarding structural interpretability, the Bacillus subtilis genome was analyzed using a two-stage dnaHNet model. The results showed that the model could spontaneously learn biologically meaningful hierarchical structures: the first stage showed sensitivity to codons and could accurately capture triplet patterns in coding regions; the second stage focused more on functional structures, with promoters, start codons, and intergenic regions showing significantly higher segmentation probabilities than coding regions.
This result indicates thatThe model not only possesses high-performance predictive capabilities, but also can reconstruct the functional organization of the genome under unsupervised conditions.This provides an interpretable computational path for parsing "DNA grammar".
Conclusion
Overall, dnaHNet no longer pre-defines sequence segmentation methods, but instead allows the model to learn them automatically. Experiments demonstrate that this dynamic, hierarchical modeling not only improves computational efficiency but also better reflects the multi-scale structure of the genome. In the long run, if it can stably learn meaningful biological units, it holds promise for revealing difficult-to-formulate patterns in the genome, opening up new avenues for research in variation prediction, functional discovery, and synthetic design.