Command Palette
Search for a command to run...
Yale University Proposed MOSAIC, Which Builds a Team of Over 2,000 AI Chemistry Experts, Enabling Efficient Specialization and Identification of Optimal Synthetic routes.
Modern synthetic chemistry faces a prominent contradiction between the rapid accumulation of knowledge and the efficiency of its application and transformation. Hundreds of thousands of related papers are published annually, and the total amount of usable synthetic knowledge has accumulated to the millions. However, most of this knowledge is scattered across different databases in the form of unstructured text, exhibiting significant fragmentation. Relying on traditional literature retrieval and manual screening is not only time-consuming and laborious, but also difficult to systematically cover reaction types across different fields. This results in a large amount of valuable information hidden in the literature being difficult to extract and transform into executable experimental protocols.
Faced with this knowledge management dilemma, the core need of synthetic practice is increasingly focused on how to efficiently obtain complete experimental procedures with high reproducibility. These procedures involve many key parameters such as reagent selection, stoichiometry control, temperature programming, and post-processing steps.
at present,The development of this field is mainly limited by two aspects.Firstly, expert experience struggles to cover the ever-expanding reaction space, often resulting in high trial-and-error costs in interdisciplinary synthetic tasks. Secondly, despite the rapid development of artificial intelligence, the application of general-purpose models in chemistry still suffers from insufficient reliability, susceptibility to "illusions," and a lack of confidence assessment, failing to meet experimental-level precision requirements. Therefore, transforming massive, fragmented chemical knowledge into structured, reliable synthetic guidance has become crucial for overcoming the field's efficiency bottlenecks.
In this context,A research team at Yale University recently proposed the MOSAIC model, which transforms a generalized large language model into a collaborative system comprised of numerous specialized chemistry experts.By effectively suppressing model illusions through professional division of labor, it provides quantifiable uncertainty assessments and realizes the systematic generation from reaction description to complete experimental protocols, which is expected to substantially improve scientific research efficiency in fields such as drug discovery and materials development.
The relevant research findings, titled "Collective intelligence for AI-assisted chemical synthesiss," have been published in Nature.

Paper address:
https://www.nature.com/articles/s41586-026-10131-4
Follow our official WeChat account and reply "MOSAIC" in the background to get the full PDF.
More AI frontier papers:
Based on the Pistachio database, we will build "AI chemistry experts" with their respective strengths.
This research was conducted using the Pistachio database, a commercial, highly structured knowledge base of chemical reactions, primarily sourced from global patent literature. Through systematic extraction and standardization of textual descriptions of reactants, products, reagents, solvents, yields, and key steps documented in the patents, the database uniformly encodes these descriptions into a machine-readable format (such as the string "SMILES").Instead of using the full dataset directly, the research team conducted a rigorous quality screening process. The core criterion was that reaction records must include detailed and actionable descriptions of experimental procedures.Rather than just the mapping relationship between reactants and products, this ensures that the model being trained learns "how to achieve the reaction" rather than just "what the reaction result is".
The filtered data was transformed into 128-dimensional reaction-specific fingerprints through a specially designed kernel metric network. This digital representation aims to capture the essential transformative features of chemical reactions, and all fingerprint vectors together constitute a "reaction universe" representing a vast space of chemical knowledge. Based on this vector space, the study employed an unsupervised Voronoi clustering algorithm (implemented using the FAISS library) to divide it into 2,489 non-overlapping specialized regions, each region clustering reaction types with highly similar chemical properties.
Ultimately, the response text within each Voronoi region was used to independently fine-tune a dedicated Llama-3.1-8B-Instruct model.This resulted in 2,489 "AI chemistry experts," each with their own strengths.The knowledge scope and capability boundaries of the entire MOSAIC framework are fundamentally determined by this patent-centric training dataset. This also explains why the system's performance is relatively limited in some rapidly developing cutting-edge fields (such as photochemistry)—these contents are not yet fully covered in existing patent databases.
MOSAIC: A decentralized collaborative system comprised of numerous professional chemical experts.
The core design idea of the MOSAIC model is to transform the general-purpose large language model Llama-3.1-8B-instruct into a decentralized collaborative system composed of numerous professional chemistry experts.This search-driven architecture significantly reduces the demand for hardware resources, requiring only a moderately sized computing configuration (such as 4 GPUs) to train specific task subsets without relying on large-scale computing clusters. The system effectively suppresses model illusions through an expert division of labor mechanism and provides quantifiable uncertainty assessments, while supporting the dynamic expansion of new experts without retraining the entire system, demonstrating significant advantages in flexibility and sustainability.
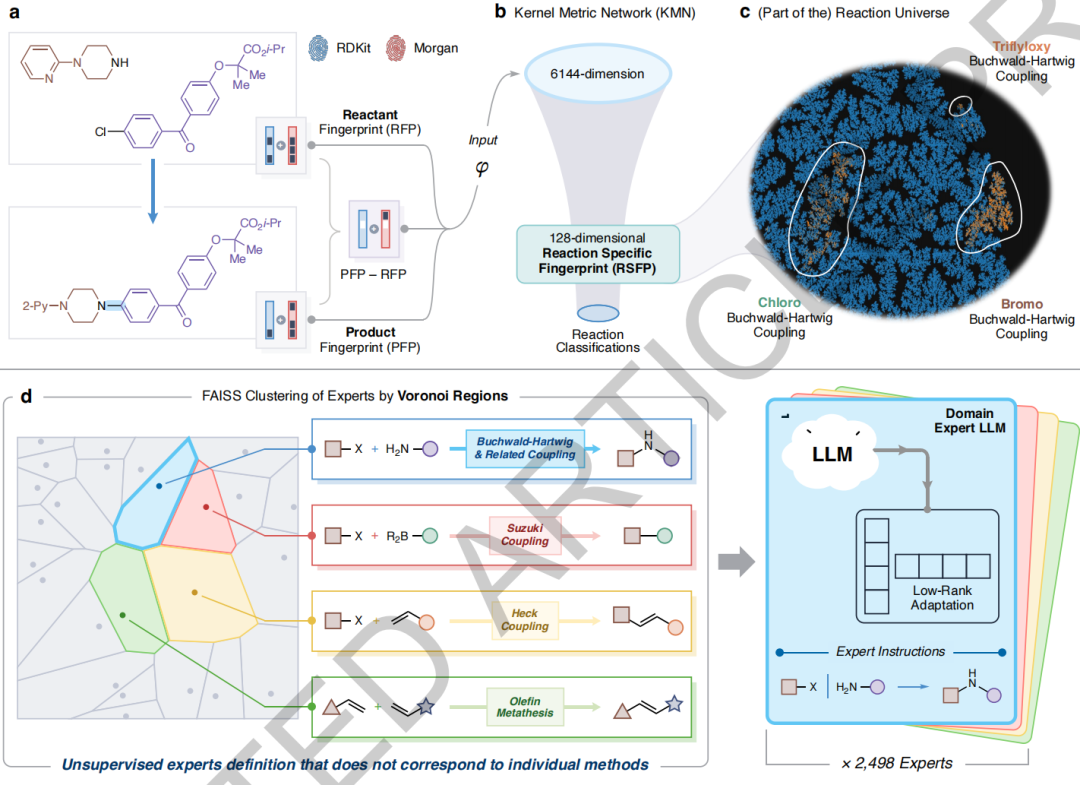
To overcome the computational and coordination bottlenecks faced by large language models when training on massive datasets, MOSAIC is built from three progressive components:
Response similarity measure:
This study designed a neural network-based nonlinear mapping (kernel metric network, KMN) to quantify the similarity between chemical reactions. It transforms reactions encoded by SMILES into 128-dimensional reaction-specific fingerprints (RSFPs), allowing their Euclidean distance to approximate reaction category relationships, thereby capturing the essential transformation features of the reactions.
Knowledge space clustering:
Leveraging the efficient indexing capabilities of the FAISS library, unsupervised Voronoi clustering is performed on the RSFP vector space, automatically dividing it into 2,498 specialized regions with highly clustered chemical properties, each region representing a specific domain of chemical knowledge.
Domain expert training:
For each cluster of reaction data, a dedicated expert model is independently fine-tuned. The study employs a two-stage training strategy: first, the basic model is fine-tuned on the complete dataset, and then the domain knowledge of the corresponding experts is deepened using the data from each cluster, enabling the experts to maintain a general understanding of chemistry while possessing profound professional knowledge.
MOSAIC first encodes the query reaction as RSFP and then uses FAISS to quickly locate its Voronoi region and corresponding expert. For example, for a Buchwald-Hartwig coupling reaction of a chloroaromatic hydrocarbon, the system will call upon an expert in this field to generate a complete and readable synthetic procedure.Experimental verification shows that, by following the procedure exactly, the target product can be obtained with a yield of 96%.
MOSAIC achieved a 94.81% TP3T component coverage and a 711% TP3T synthesis success rate.
This study further validated the comprehensive performance of the MOSAIC model through a multi-dimensional evaluation system. Its core value lies in transforming massive amounts of literature knowledge into highly reliable synthetic intelligence.
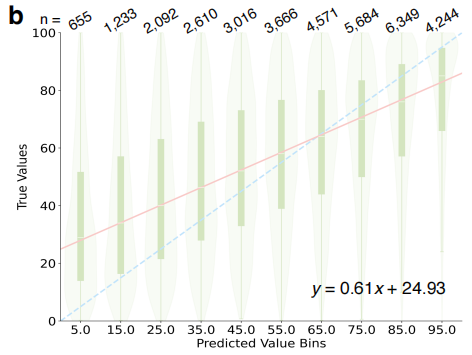
In terms of yield prediction and core component identificationThe MOSAIC model achieves quantitative prediction of reaction yields by parsing the complete experimental procedure text. As shown in the figure below, after adopting the binning strategy, the center of the predicted interval shows a significant correlation with the median of the actual yield (R² = 0.811). The model demonstrates excellent coverage in identifying key reaction components (reagents, solvents).After integrating the predictions of the top three experts, the overall success rate of identifying at least some of the correct components is as high as 94.8%.It is worth noting that even if the prediction conditions are not entirely consistent with the literature records, the output is often a chemically feasible alternative, reflecting a deep level of professional judgment.
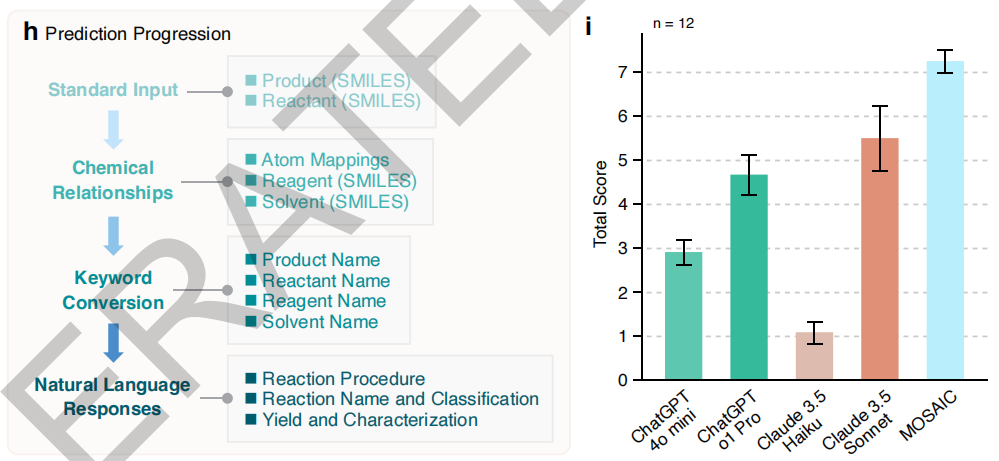
In comparative tests of 12 important reactions (Suzuki coupling, Buchwald-Hartwig amination, etc.), as shown in the figure below, MOSAIC consistently outperforms general-purpose models such as ChatGPT-4o and Claude 3.5 in providing clear and feasible synthetic guidance. This advantage is particularly evident given that the model has only 8 billion parameters, demonstrating the effectiveness of domain-specific fine-tuning. More importantly,MOSAIC overcomes the problems common to general models in chemistry tasks, such as unstable instruction compliance and arbitrary responses, and provides stable and reliable output.This is crucial for actual experiments.
To assess the practicality, versatility, and reliability of the proposed framework, this study also conducted extensive experimental validation by performing precise, top-ranking predictions of fundamental reactions in modern chemical synthesis. The researchers focused on broadly applicable catalytic reactions crucial for drug and materials development. The carbon-nitrogen bonds formed by Buchwald-Hartwig aminations are ubiquitous in drug molecules, and the conditions for these challenging reactions were accurately predicted. Efficient assembly of drug-grade scaffolds was achieved, demonstrating particular advantages in olefin transformations crucial for applications ranging from natural products to functional materials.
Furthermore, the practicality of the MOSAIC model has been strongly demonstrated in the successful synthesis of a large number of novel compounds.Of the 37 target compounds synthesized, 35 were successful on the first recommendation of the model, with an overall success rate of 71%.The validation scope covers everything from classical coupling reactions to selective transformations, and includes an innovative case study demonstrating the ability to guide the development of novel azaindole cyclization methods.
Most importantly, the confidence index (distance to the nearest expert centroid) within the model shows a clear positive correlation with the experimental success rate: the success rate of high-confidence predictions (distance < 100) exceeds 75%. This provides chemists with valuable quantitative decision-making support, enabling them to effectively allocate resources between high-success-rate goals and exploratory attempts.
Chemical synthesis enters a new era of precision intelligent manufacturing
In the global process of promoting intelligent chemical synthesis, academia and industry are working together along complementary paths to reshape the entire chain from molecular discovery to process production.
University research is like pioneering exploration of the unknown, focusing on overcoming the limits of underlying computing and innovating scientific research paradigms.Researchers at MIT have cleverly transferred the "diffusion model" used for image generation to the field of chemical reactions.It achieves ultra-fast computation of key "transition state" structures—compressing tasks that would typically take days to complete into seconds, and providing unprecedented microscopic insights into reaction predictions with atomic-level precision of 0.08 angstroms.
Meanwhile, the Stanford University team is dedicated to reshaping the way research itself is conducted.The system builds an AI-driven "virtual laboratory" capable of autonomously forming multidisciplinary virtual teams.Coordinated by the "lead researcher AI," collaboration and debates are conducted in seconds, resulting in innovative ideas that transcend conventional approaches in complex topics such as vaccine design. Furthermore, research from institutions like Harvard University has pushed the simulation capabilities of artificial intelligence to a macroscopic scale. Their proposed unified framework has successfully achieved precise simulations of complex ferroelectric materials containing millions of atoms, providing a powerful digital lens for fundamentally designing next-generation functional materials.
Compared to academia's pioneering spirit, corporate innovation focuses more on translating cutting-edge algorithms into productivity and market competitiveness that solve real-world pain points. German chemical giant BASF has deployed AI globally, launching not only "AI Chemist Copilot" to assist in research and development, but also...The development cycle of new materials has been significantly shortened by 60%.Furthermore, AI is deeply integrated into production optimization, logistics planning, and predictive maintenance, achieving efficiency improvements across the entire value chain from laboratory to factory.
In the pharmaceutical field, companies like Novartis, headquartered in Switzerland, are embracing AI in an "end-to-end" manner. Through in-depth collaborations with professional companies such as Isomorphic Labs and Schrödinger, they are applying artificial intelligence to every key stage, from the discovery of new targets, compound generation and safety prediction, to the optimization of clinical trial design, significantly improving the certainty and success rate of drug development.
Looking at these breakthroughs spanning academia and industry, chemical research—a traditional discipline that once heavily relied on personal experience and repeated trial and error—is being profoundly reshaped by data and algorithms, steadily moving towards a new era of precise science that is predictable, plannable, and automated. From innovative drugs that conquer diseases to green materials that contribute to sustainable development, this far-reaching transformation of intelligent chemical synthesis is forging unprecedented core capabilities to help us address the most pressing challenges of our time.
Reference articles:
1.http://edu.people.com.cn/n1/2025/0730/c1006-40532541.html
2.https://cen.acs.org/pharmaceuticals/drug-development/Q-Novartiss-biomedical-research-head/103/web/2025/01








