Command Palette
Search for a command to run...
With Computational Costs Halved, ChemOntology, a Chemical Reaction Discovery Tool, "encodes" Human Intuition Into Its System, Accelerating the Search for Reaction pathways.
Chemical reaction mechanisms not only reveal the intrinsic laws governing the transformation of matter, but also provide crucial evidence for industrial applications such as the design of efficient catalysts and the development of green synthesis pathways. Analyzing reaction mechanisms relies heavily on a key computational technique—reaction pathway search—which involves locating local minima and reaction intermediates on the potential energy surface (PES) to help map the actual reaction pathway.
For a long time, computational chemists have relied primarily on intrinsic reaction coordinates (IRC) methods to explore reaction mechanisms by generating finite configurations. However, this traditional approach has significant limitations. It is often constrained by the researcher's pre-set path and is prone to overlooking unconventional reaction pathways, thus potentially missing potential alternative mechanisms.
With the development of automated methods such as Artificial Force-Induced Response (AFIR), unbiased response path search has become possible. These methods treat response paths as a network of interconnected "nodes," and systematically explore response possibilities by iteratively generating new configurations, thus opening a new window for discovering unknown response mechanisms.
However, automated pathfinding is not a perfect solution. The energy calculations for numerous configurations incur high costs, and the necessity of studying the mechanisms underlying conformational changes further exacerbates the computational burden. Although semi-empirical methods and machine learning potential functions can partially reduce costs, occasional inaccuracies in energy prediction can still affect the reliability of pathfinding.
Chemical ontology, as a "knowledge structuring tool," offers a new approach to overcoming the aforementioned bottlenecks. Through standardized definitions of entities, attributes, relationships, and rules, it organizes fragmented chemical knowledge into machine-readable and processable structured information. For example, ontology frameworks such as RXNO have already demonstrated their value in reaction pathway annotation.
Building on this, a research team at Hokkaido University in Japan has developed a novel AI system, ChemOntology.As a chemical knowledge classification system, it formalizes human chemical reasoning into a machine-understandable framework, thereby enabling rapid exploration and analysis of chemical reactions.The successful application of this framework in the study of the classic Heck reaction mechanism not only verifies its effectiveness in accelerating path search, but also highlights the enormous potential of integrating "human chemical knowledge" with "automated computation".
The related research findings, titled "ChemOntology: A Reusable Explicit Chemical Ontology-Based Method to Expedite Reaction Path Searches," have been published in ACS Catalysis.
Research highlights
* It successfully "programs" the intuition of human chemists into the system without relying on a training dataset, which is a significant advantage over traditional machine learning methods;
* Experimental results show that, when combined with AFIR, ChemOntology can achieve results comparable to a full AFIR_TARGET search when exploring about half the number of paths, reducing the overall computational cost by nearly half.
Paper address:
https://pubs.acs.org/doi/10.1021/acscatal.5c06298
Follow our official WeChat account and reply "ChemOntology" in the background to get the full PDF.
More AI frontier papers:
https://hyper.ai/papers
Data Methodology of Knowledge-Driven Framework
The data resources relied upon by this research institute are not the massive datasets traditionally used to train machine learning models. This is precisely due to the inherent characteristics of ChemOntology as a knowledge-driven framework: it focuses on chemical rules and mechanisms rather than relying on data fitting, thus avoiding the high dependence on large-scale data and its potential limitations at the methodological level.
first,Researchers used the public chemical database PubChem to obtain standardized information on all key components in the reaction.This includes molecular structure, name, and unique identifier. This information can be considered as an "identity card" for each chemical substance, which not only helps to accurately define the role of each component in the reaction system, but also allows for the tracking of target products and the elimination of irrelevant or unnecessary byproducts through unique compound numbers, thereby making the search for subsequent reaction pathways more accurate and efficient.
Secondly, in order to test the reliability and applicability of the method in real and complex chemical scenarios,The researchers selected the classic Heck reaction, which has diverse mechanisms and numerous reaction steps, as a test case.The system was provided with complete input information, including three-dimensional structural files of reactants, catalysts, ligands, and bases, as well as reference energy data for known intermediates and final products. This representative case fully examines the method's performance in complex reaction networks, not only verifying its ability to identify key intermediates and distinguish between main and side reaction pathways, but also intuitively demonstrating its advantages in reducing computational costs.
Overall, this study ensures the accuracy of information through authoritative databases, verifies the effectiveness of methods by leveraging typical complex reactions, and promotes collaboration and iteration through full open source, enabling it to maintain broad applicability to diverse organometallic reaction systems without relying on large-scale training data.
ChemOntology: A New Framework for Pathway Search in Organometallic Reactions
ChemOntology is a knowledge-driven computing framework whose core idea does not rely on training models with large-scale data.Instead, it systematically integrates chemical reaction rules, structural constraints, and quantum chemical pathway search processes.This allows for efficient exploration of reaction pathways within a defined chemical context. The method uses AFIR (Artificial Force Induced Reaction) as its computational engine, explicitly encoding chemical knowledge to guide the search direction and performing real-time screening of generated structures to avoid meaningless or unreasonable reaction evolutions.
As shown in the figure below, the ChemOntology workflow consists of user input parsing, chemical information modeling in setup file, reaction path generation using ERPOs, structural rationality constraints, running and controlling AFIR, and path analysis.
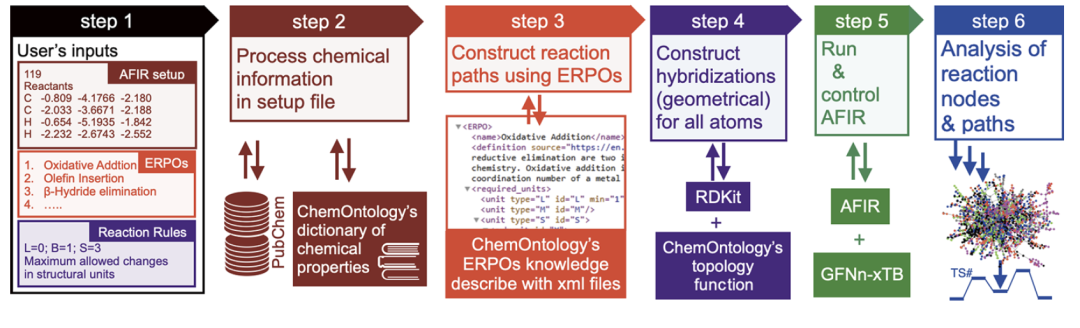
The reaction system is first analyzed as a collection of structural units such as metals, ligands, substrates, and optional bases, with each type of unit assigned a specific chemical role and property.The reaction process is described as a gradual transformation of the hybridization states of structural units and their internal atoms, thus tracing structural changes at three levels: "reaction node—structural unit—atom". This hierarchical representation allows the model to determine the chemical plausibility of reaction paths based solely on geometric and topological information, without relying on details of electronic structure.
The generation of reaction pathways relies on ERPO (Elementary Reaction Pathway Operator).That is, a modular description of common organometallic elementary reaction processes.Examples of reactions include coordination compound formation, oxidative addition, olefin insertion, and β-hydrogen elimination. ERPO is not only used to construct reaction sequences but also serves as a rule-verifying tool during the search process, ensuring that each structural transformation conforms to the expected chemical semantics. By breaking down complex reactions into combinatorial elementary processes, ChemOntology can significantly reduce the combinatorial complexity of the search space while maintaining reaction diversity.
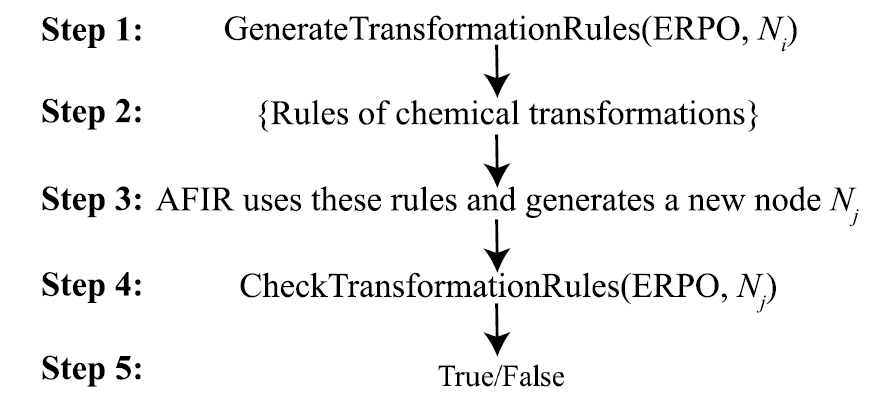
To further constrain reaction evolution,ChemOntology introduces a filtering mechanism based on changes in atomic hybridization.Users can limit the maximum allowable structural adjustments for different structural units throughout the reaction process using a few parameters. Geometric structures exceeding the constraints are automatically identified and removed from the search. This mechanism effectively suppresses the structure explosion problem and significantly improves computational efficiency without pre-setting specific reaction outcomes.
In practical computation, ChemOntology is embedded as a knowledge control layer above the AFIR search process, combined with the semi-empirical tight-binding method GFN2-xTB to describe the geometric evolution of the reaction path. Unlike machine learning models,ChemOntology does not require training with a dataset; its "knowledge base" mainly consists of functional group recognition rules, structural unit classification schemes, and ERPO files.All of these can be flexibly modified by the user according to the research object. This design makes ChemOntology more like a computational chemical methodology, used to systematically introduce human chemical intuition into the automated reaction exploration process.
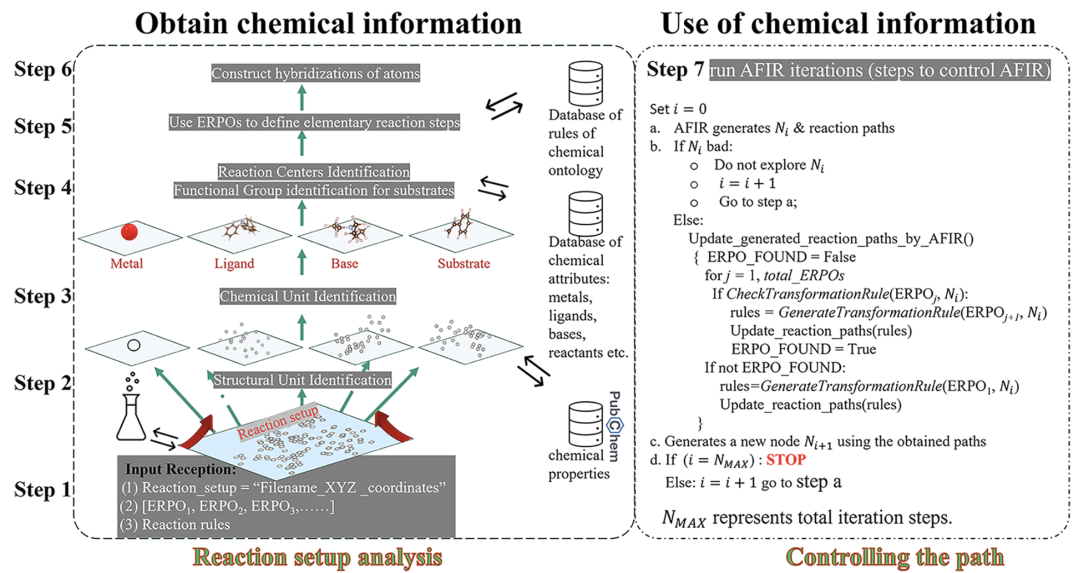
Overall, ChemOntology provides a platform for searching reaction pathways under explicit chemical constraints: it does not restrict the emergence of new reactivity, but rather guides computations to explore within a "reasonable chemical space" through structured rules, thereby achieving a balance between reaction mechanism analysis and potential new chemical discoveries.
Experimental results: Computational cost halved, path clarity doubled.
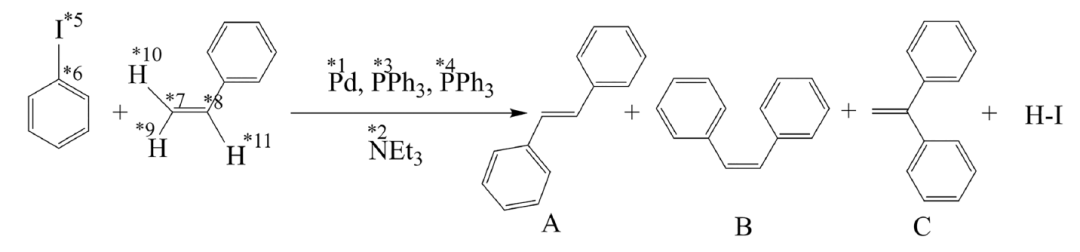
To verify the effectiveness and efficiency of the ChemOntology framework in reaction pathway search,The research team selected the classic Heck reaction, which has a complex mechanism and is representative, as the test system.As shown in the figure below, this reaction uses iodobenzene and styrene as substrates. Under palladium catalysis, triphenylphosphine ligand, and triethylamine base conditions, it mainly produces trans-stilbene, accompanied by a small amount of cis isomer and trace byproducts. Its mechanism involves multiple key steps, including oxidative addition, olefin insertion, migratory insertion, β-hydrogen elimination, and base elimination. The numerous reaction centers pose a typical challenge to automated pathfinding methods.
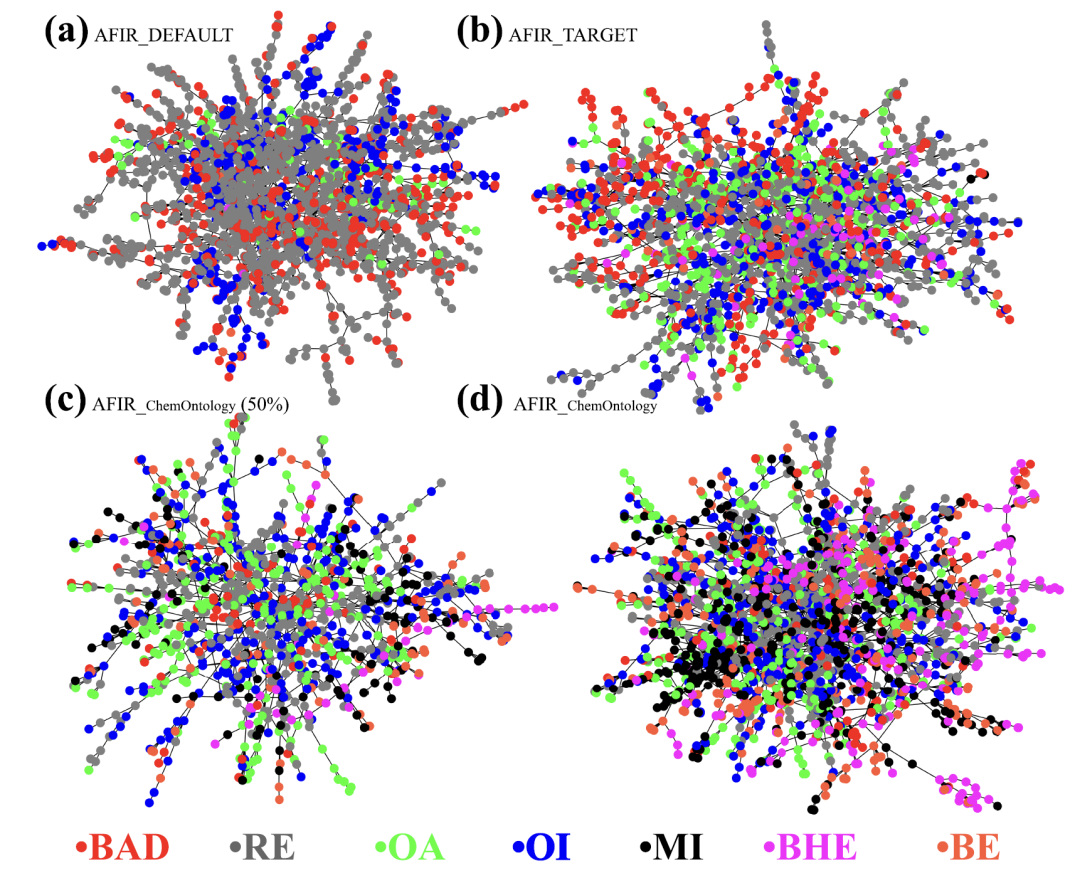
The study compared three parallel path search strategies: the unguided AFIR_DEFAULT, the partially constrained AFIR_TARGET, and the AFIR_ChemOntology, which incorporates chemical ontology. These three strategies differ fundamentally in their level of "intelligence": the former traverses the configuration space almost indiscriminately, while the latter narrows the search scope through artificial constraints.AFIR_ChemOntology, on the other hand, automatically identifies the chemical roles of reactants and key reaction centers through its framework, and dynamically guides the search by leveraging elementary reaction processes.
Under the same computational conditions, as shown in the figure below, the reaction networks generated by the three methods differ significantly. AFIR_DEFAULT produces a large number of chemically meaningless invalid nodes, severely flooding effective paths; AFIR_TARGET, while showing some improvement, still has many redundant structures; in contrast,The search results for AFIR_ChemOntology are highly focused, enabling the early and clear identification of major reaction pathways.The calculations were focused on chemically plausible pathways. Further intermediate statistics showed that ChemOntology significantly reduced the proportion of "bad nodes," and the identified key intermediates were highly consistent with the classical mechanism of the Heck reaction.
As shown in the figure below, energy analysis reveals that all three methods capture a common step in the early stages of the reaction.However, only AFIR_ChemOntology can completely distinguish and track the specific pathways leading to the main product and the byproduct respectively.Furthermore, characteristic interactions associated with β-hydrogen elimination were generally observed in the efficient pathway, while in the pathway leading to trace products, these interactions exhibited weaker structural stability, which may explain their lower generation probability.
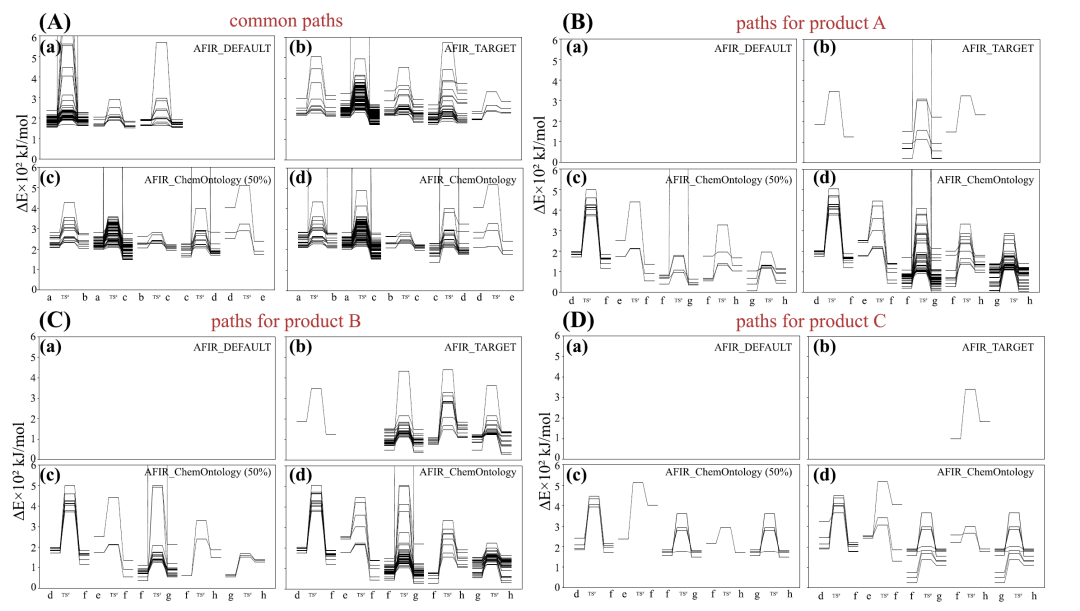
In terms of computational efficiency,AFIR_ChemOntology achieves comparable efficiency to a full search of AFIR_TARGET while exploring about half the number of paths, reducing overall computational cost by nearly half.This advantage primarily stems from the guidance of chemical knowledge in the search direction and the real-time filtering of invalid structures. Overall, the experimental results demonstrate that integrating chemical ontology into automated path search can significantly improve the efficiency of mechanistic analysis while ensuring chemical rationality, providing a more efficient and reliable approach for the study of complex reaction systems.
From Laboratory to Factory: Reshaping the Path of Reaction Exploration Through Chemical Ontology
The integration of chemical ontology and automated reaction pathway searching is building a crucial bridge connecting theoretical chemistry and industrial applications. This trend has not only spurred a series of cutting-edge explorations in academia but has also triggered substantial innovative practices in industry, driving the transformation of reaction mechanism research from traditional "post-hoc analysis" to more proactive "active guidance."
In academia, research focuses on algorithmic innovation and mechanism refinement, continuously expanding the boundaries of knowledge in this field. For example, a team at the University of Iceland developed the "Optimal Transport Gaussian Process" (OT-GP) algorithm.Its core lies in adopting an intelligent data filtering strategy, which can work efficiently using only a fixed amount of training data.This algorithm significantly reduces the average time for molecular reaction path search from 28.3 minutes to 12.6 minutes, and significantly improves the success rate, providing a new tool for rapid mechanism exploration of complex systems.
Paper title: Adaptive Pruning for Increased Robustness and Reduced Computational Overhead in Gaussian Process Accelerated Saddle Point Searches
Paper link:https://doi.org/10.48550/arXiv.2510.06030
at the same time,A research team at ETH Zurich in Switzerland combined ab initio molecular dynamics with enhanced sampling methods.We systematically studied the key hydrogen transfer and rearrangement steps in the catalytic reaction of molecular sieves and transition metals, revealed the mechanism characteristics of the dynamic changes of reaction channels with the reaction environment, and proposed a general microscopic picture that can be used to guide the rational design of catalysts.
Paper title: Ab initio molecular dynamics with enhanced sampling in heterogeneous catalysis
Paper link:https://pubs.rsc.org/en/content/articlelanding/2022/cy/d1cy01329g
Industry practice, on the other hand, focuses more on translating these theories into practical productivity. Take Schrödinger, a representative company in the field of computational chemistry in the United States, as an example.Its AutoRW automated reaction workflow deeply integrates the structured thinking of chemical ontology.It achieves full-process automation from reaction enumeration and path mapping to result organization and output.
Meanwhile, the collaboration between German chemical giant BASF and IBM also demonstrates a similar path of technological integration.Both parties will combine chemical ontology with quantum chemical calculations and artificial intelligence to jointly tackle the research and development of high-performance catalysts.By adopting the "knowledge-guided + AI computing" model, not only has the R&D cycle been significantly shortened and the cost of experimental trial and error been reduced, but a solid foundation has also been laid for the application of polyurethane materials in the automotive, construction and other fields.
These practices from leading global companies not only validate the universal value of combining chemical ontology with automated computing, but also, through cross-regional and cross-domain technological collaboration, have formed a virtuous cycle from academic breakthroughs to technology transfer, and then to industrial applications and demand feedback, continuously driving the global chemical industry toward a greener, more efficient, and smarter future.
Reference Links:
1.https://wp-stg.schrodinger.com/wp-content/uploads/2023/10/A4-22_111-Reaction-Workflow-Application-Note_R3-1-1.pdf
2.https://blog.csdn.net/cainiao080605/article/details/147259567
3.https://phys.org/news/2025-12-ai-mimics-human-intuition-explore.html








