Command Palette
Search for a command to run...
Multimodal Models Accelerate the Matching of New Materials With Industrial Applications, Predicting Material Properties Without the Need for a Complete Crystal structure.
Oranges grown south of the Huai River become oranges, while oranges grown north of the Huai River become tangerines. The same seed planted in different soils and climates yields vastly different fruits. This natural law of growth is equally evident in materials chemistry—new materials exhibit varying performance for different applications. Research indicates that scientists create hundreds of thousands of new materials every year. These are like countless "seeds" of immense potential that need to be rooted in a suitable environment to thrive.
Although current new material designs are often synthesized for specific applications, they often have potential uses in different fields. However, how to quickly determine the application scenarios of new materials remains a challenging task.Take the widely used crystalline materials metal-organic frameworks (MOFs) as an example. Their most notable application is as storage media for gases such as hydrogen and methane, and they demonstrate excellent performance potential in membranes, thin-film devices, catalysis, and biomedical imaging. Traditional approaches to determining the optimal application of MOFs rely on material properties as an intermediate basis, but testing is costly (in terms of time, equipment, and expertise). Furthermore, computational screening and machine learning methods require the complete crystal structure to predict properties, but crystal structure analysis is time-consuming and not readily available after MOF synthesis.
To address this, a research team from the Department of Chemical Engineering and Applied Chemistry at the University of Toronto, Canada, proposed a new method based on a multimodal machine learning model.Using information available after MOF synthesis to predict their potential properties and uses,For example, the model includes powder X-ray diffraction patterns (PXRD) and the chemicals used in its synthesis. The research team added an application recommendation system to the model, which provides application suggestions immediately after MOF synthesis. This research accelerates the connection between the synthesis of metal-organic frameworks (MOFs) and their application scenarios.
The related research was published in Nature Communications under the title "Connecting metal-organic framework synthesis to applications using multimodal machine learning."
Research highlights:
* This method uses only information available after synthesis to predict the potential properties and uses of MOFs, making application recommendations immediately after the MOFs are synthesized, significantly shortening the material synthesis to application cycle;
* The model's prediction performance is comparable to that of advanced models that require precise crystal structure input (e.g., CGCNN and MOFormer), and even outperforms them under certain conditions. It is also stable and reliable in the face of experimental noise, crystal structure defects, and other conditions, demonstrating excellent robustness.
* This study combines a visual application recommendation system to build a synthesis-prediction-application closed-loop system;
Paper address:
https://www.nature.com/articles/s41467-025-60796-0
Follow the official account and reply "MOFs" to get the full PDF
More AI frontier papers: https://hyper.ai/papers
"Data is the synthesis site": MOFs data construction strategy for application prediction
In this study, a total of 6 metal-organic framework (MOFs) databases were used for model training and evaluation: CoRE-2019, BW20K, ARABG, QMOF, hMOF, and CSD subsets.in:
* hMOF provides an extremely large library of hypothetical structures, which helps improve the generalization ability of the model.
* BW20K and ARABG are used to enhance diversity and support few-shot tasks.
* The CSD subset is used to test the robustness of the model under experimental bias.
The research team used crystal structures from the CoRE 2019, BW20K, ARABG, QMOF, and hMOF databases, and calculated simulated PXRD patterns from 0 to 90 degrees using the pymatgen XRD module to simulate the structural characterization information obtained after synthesis in actual experiments. Chemical precursor information, consisting of metal nodes and organic linkers, was constructed in the format of [metal type].[organic linker], which was input into the model's Transformer channel for word segmentation.
A multimodal learning framework driven by self-supervised pre-training
The research team proposed a self-supervised pre-training driven multimodal learning framework, aiming to get rid of the dependence on the complete crystal structure.Predict the properties and potential applications of MOFs using only information available post-synthesis.
The workflow of this self-supervised multimodal model is shown in the figure below. A precursor string and a powder X-ray diffraction (PXRD) spectrum are used as inputs, embedded through a Transformer and a convolutional neural network (CNN), respectively, and passed to a regression head for fine-tuning. The precursor provides information about the material's chemical properties, while the PXRD pattern provides additional information about the overall geometric structure.
The chemical precursor strings encoded by Transformer and the PXRD spectra processed by CNN are constructed into a unified representation space through feature splicing and projection.To make up for the deficiency of “precursor + PXRD” in being unable to directly characterize the local chemical environment, the research team introduced a self-supervised pre-training mechanism.The model output is aligned with the embedding of the crystal graph convolutional neural network (CGCNN), and the cross-correlation matrix is constrained to be close to the identity matrix through the Barlow Twins loss, thereby guiding the model to learn the expressive power of the local chemical environment.
On this basis, after self-supervised training on large-scale unlabeled data, the model can converge quickly with limited labeled samples and achieve high-precision predictions of pore structure, chemical dependence characteristics and quantum chemical properties.
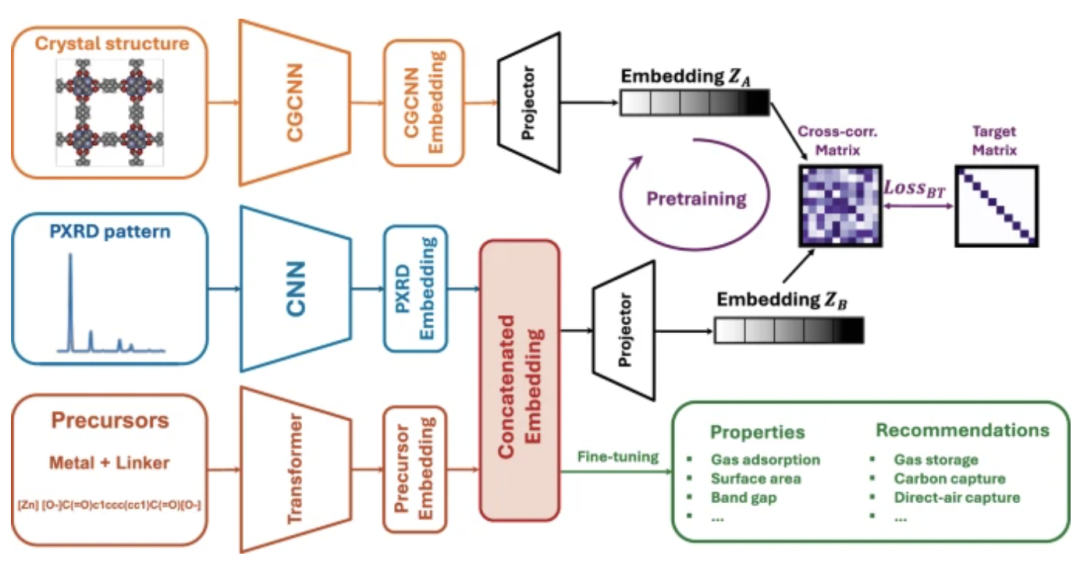
Specifically, based on the crystal structures obtained from the MOFs database,Even with small amounts of data, this method can accurately predict various characteristics.These include pore structure, chemically dependent properties, and quantum chemical properties.
During the self-supervision and training phase, a self-supervised learning (SSL) pipeline was constructed to perform representation learning between the Crystal Graph Convolutional Neural Network (CGCNN) and the model, overcoming the model's limitation of being unable to understand the MOF's local environment from its input. The model's weights were initialized, enabling rapid convergence to a solution. Self-supervised learning was performed on the CGCNN embeddings. Each embedding of size 512 was extracted from the CGCNN and the model's projector, and a cross-correlation matrix of shape (512, 512) was constructed. The Barlow-Twin loss function was used to minimize the difference, bringing the cross-correlation matrix close to the identity matrix, thereby achieving representation learning.
Evaluation of multimodal models
To prove that the model can effectively predict various MOFs properties and lay the foundation for the combination of MOFs synthesis and application, the research team evaluated the accuracy of the model, using the Spearman rank correlation coefficient (SRCC) and mean absolute error (MAE) to evaluate the model's prediction accuracy in geometrically dependent properties, chemically dependent properties, and quantum chemical properties, and conducted benchmark comparisons with CGCNN, MOFormer, and descriptor-based machine learning models.
The results show thatThe model accuracy of this model is comparable to that of models relying on the complete crystal structure.It even outperforms CGCNN and MOFormer in geometric performance, thus verifying that high-accuracy property prediction can be achieved using only synthesis information, laying an experimental foundation for the rapid matching of MOFs synthesis to application.
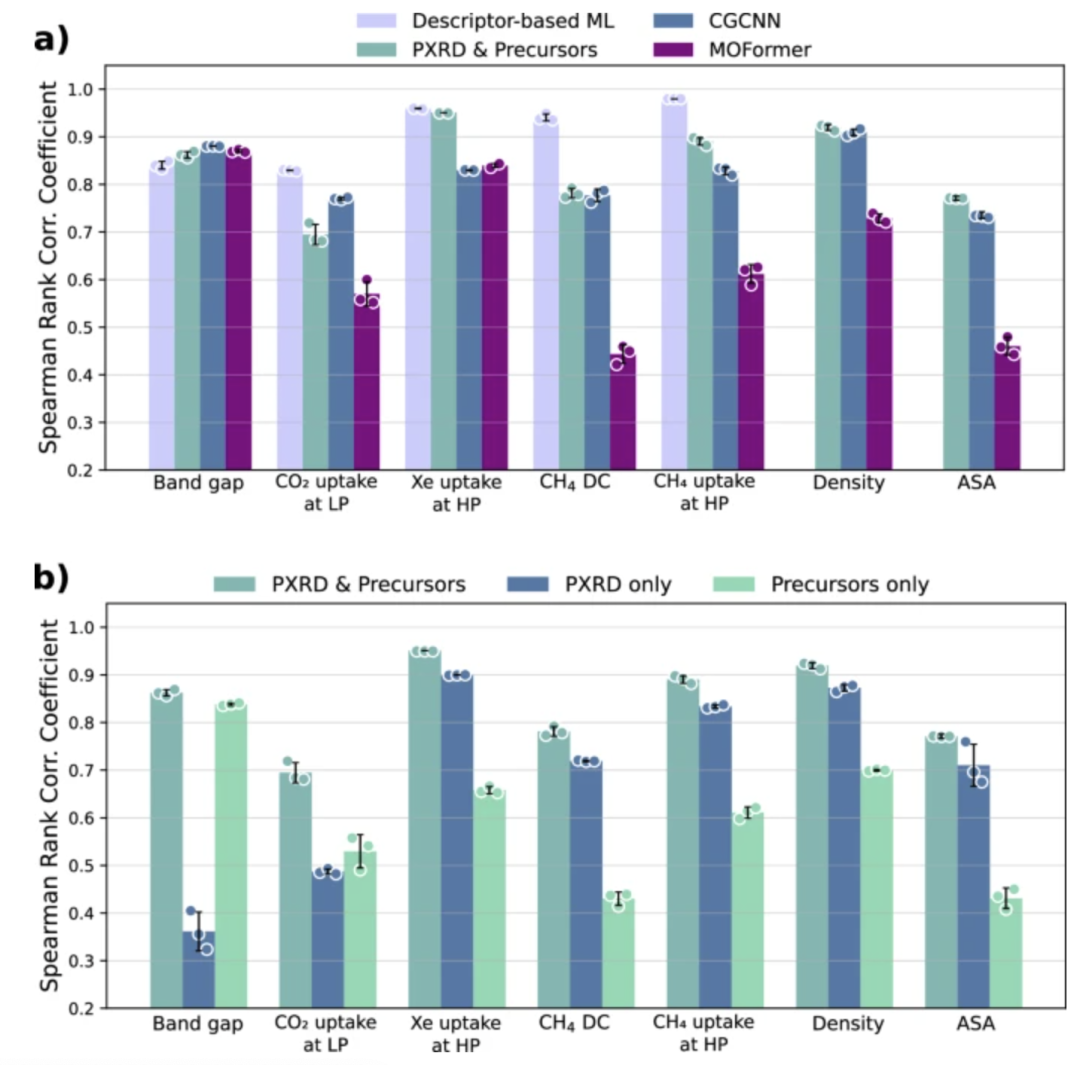
In addition, the team conducted ablation experiments, comparing models that relied solely on chemical precursors and models that relied solely on PXRD with the multimodal model developed in this study. The results showed that the model that accepted only chemical precursors as input failed to effectively capture the overall MOF structure, scoring poorly on geometry-related and purely geometric properties. While the model that accepted only PXRD captured the overall MOF structure well, it failed to reflect the local environment, resulting in poor scores on chemical-related and quantum chemical properties (such as CO₂ adsorption at low pressure and band gap). Both models exhibited limitations. The results demonstrate that only by combining PXRD (which provides geometric information) with the precursor string (which provides chemical information) can the multimodal model achieve comprehensive and accurate predictions for all three property categories; using either modality alone clearly underperforms.
Model stability verification: robustness assessment against structural errors and experimental noise
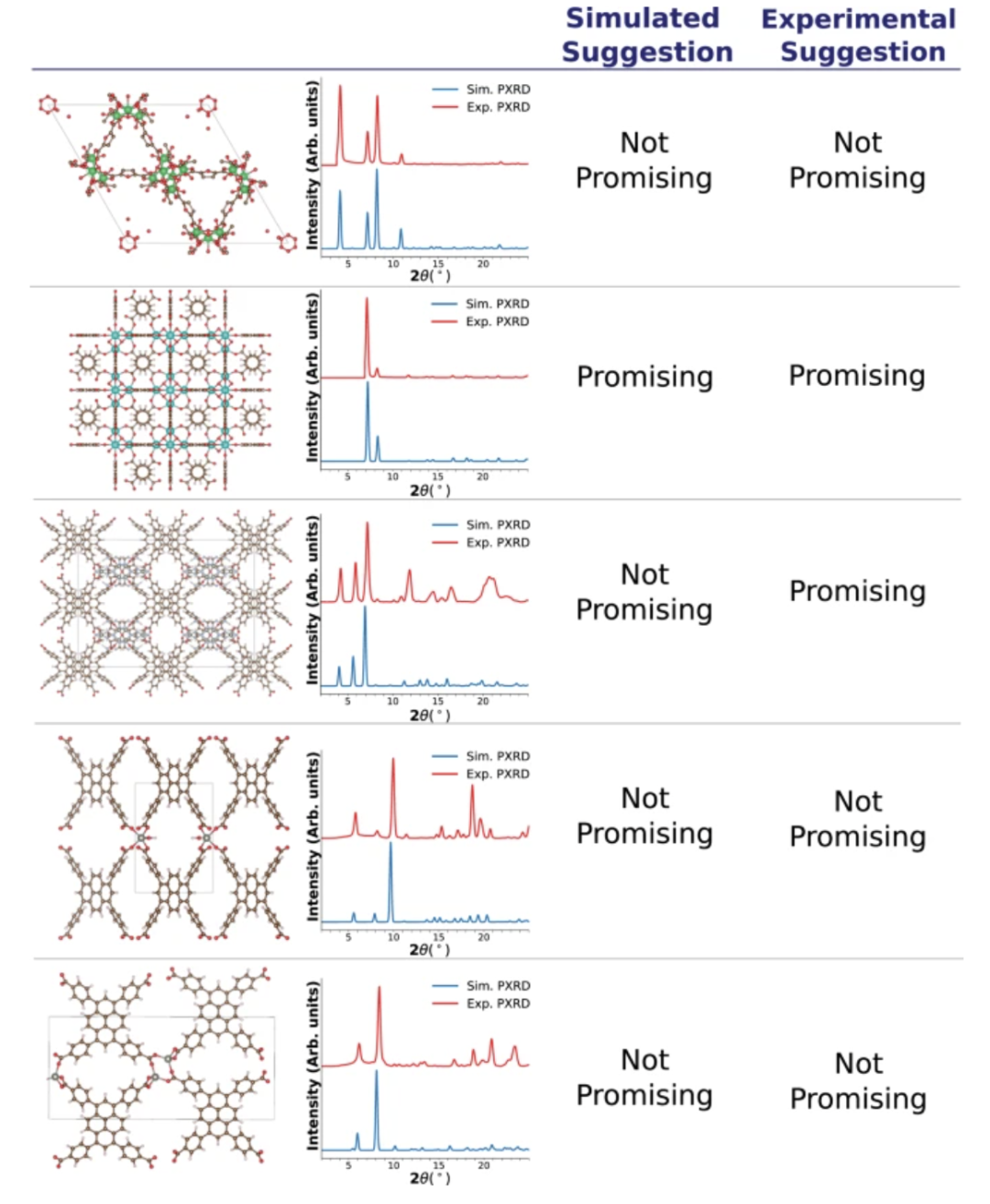
Stability is an important metric for evaluating whether a machine learning model can be reliably applied in real-world scenarios. To this end, the research team systematically evaluated the robustness of the proposed multimodal model under non-ideal conditions. First, the researchers used experimental crystal structures extracted from the Cambridge Structural Database (CSD) to calculate the corresponding PXRD patterns to simulate common structural deviations in real experiments, such as missing hydrogen atoms and the presence of bound or unbound solvent. The evaluation focused on a single geometry-related property: the prediction of methane adsorption capacity under high pressure for methane storage applications.
The results show thatThe model can still maintain good predictive ability under the above variation conditions.The ranking of CH₄ high-pressure adsorption performance has strong consistency, and the relative error is controlled below 13%, showing high robustness.
On this basis, the team further introduced actual measured PXRD patterns for testing to verify the stability of the model in the face of actual measurement errors such as instrument noise and temperature fluctuations. Although there are significant differences between the simulated and experimental patterns in some samples, the model still gives recommended results close to the simulated patterns in most cases, and only differs in individual cases with significant noise or obvious peak misalignment. Combined with the above experiments, it shows that the multimodal model not only has a high degree of prediction accuracy under ideal structural input conditions, but also maintains robust performance when the experimental structure is imperfect or there is noise in PXRD.Its wide applicability in practical material research and applications has been verified.
The figure below shows the model recommendation results, comparing the differences between the simulated PXRD patterns and the experimental PXRD patterns:
Synthesis-application integrated recommendation system
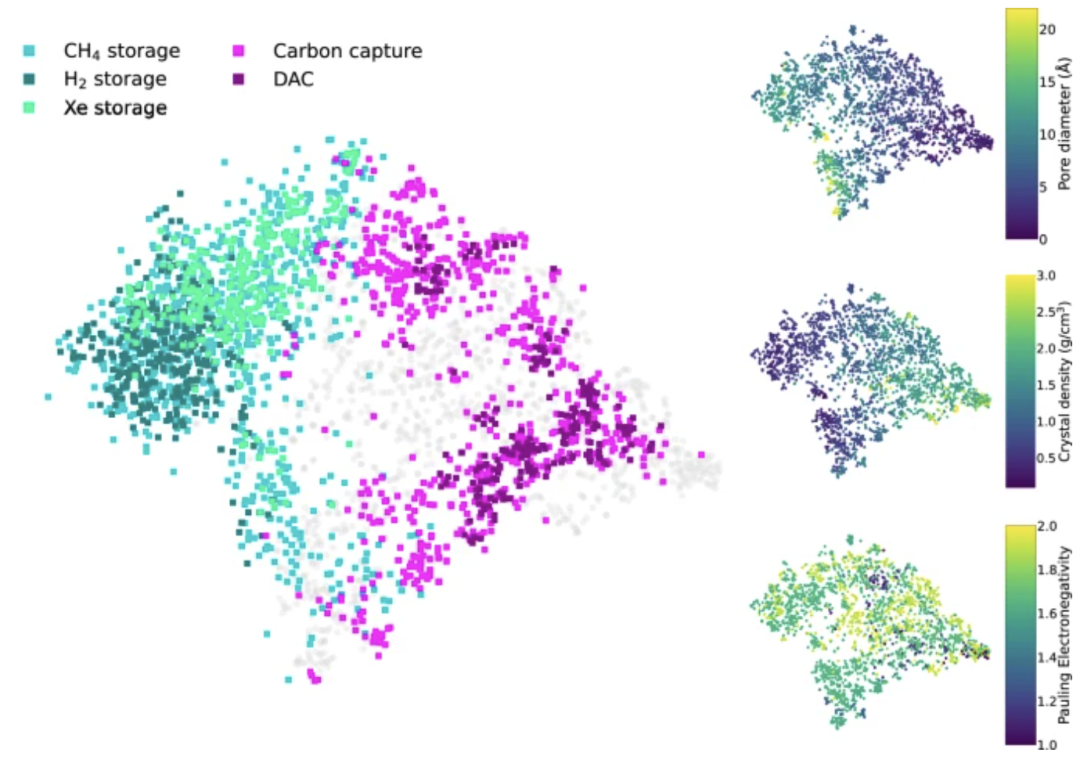
Based on the model's impressive performance, researchers developed a visual potential application recommendation system that matches newly synthesized MOFs with potential applications (such as gas storage and carbon capture) based on predicted material properties. Using t-SNE technology, the system projects the modal model's latent space and uses color to represent the recommended applications for MOFs. The following figure illustrates the mapping of synthesis information to application scenarios:
To verify the model's ability to predict future material applications, the researchers conducted a time-travel experiment.The model was trained using CoRE-2019 entries stored in the CSD database before 2017, and tested using entries stored after 2017 to simulate predictions for future materials. The goal of the experiment was to predict the performance of these MOFs in a specific application: carbon dioxide adsorption. The results showed that the model successfully identified 18 MOFs with potential for carbon capture, 15 of which were originally designed for other applications.
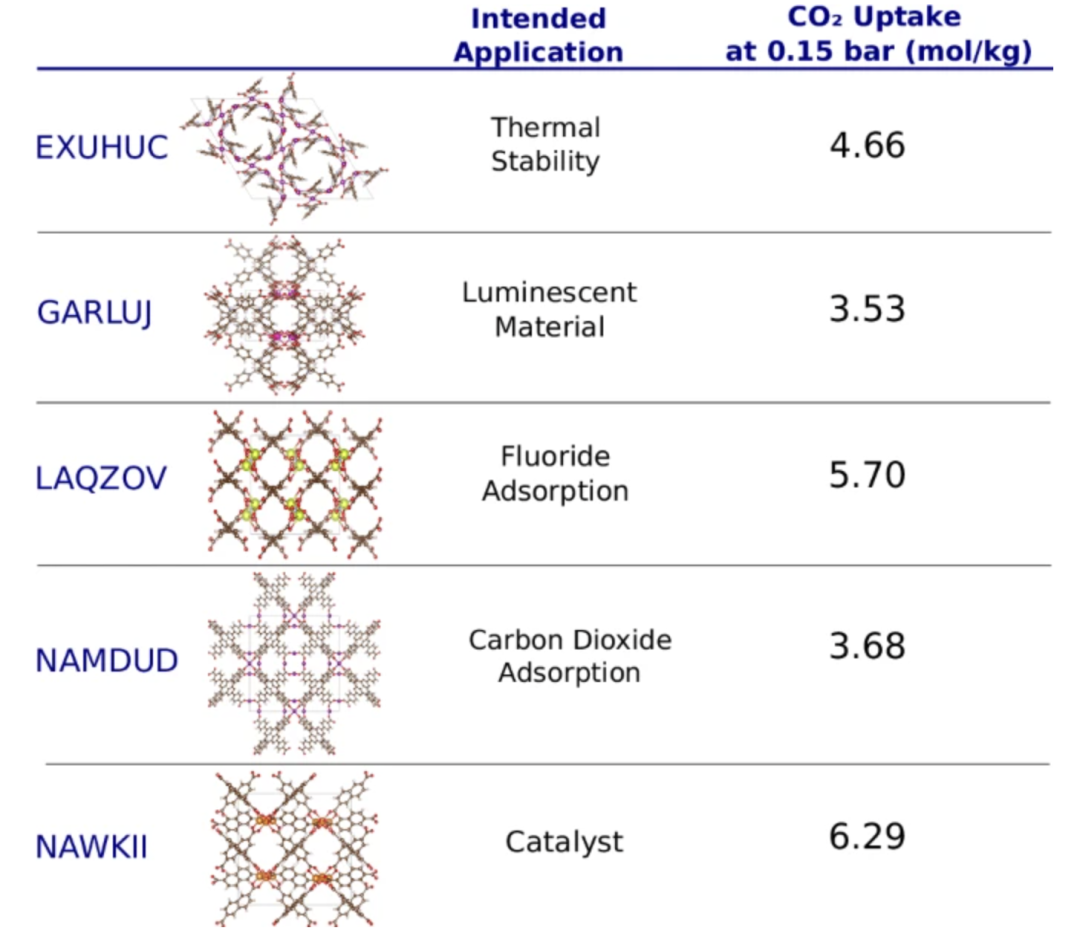
Some of these MOFs and their expected synthetic applications based on the corresponding
Machine learning is revolutionizing materials science
This article introduces a multimodal machine learning approach that accurately predicts the diverse properties of MOFs and matches them to specific applications, regardless of crystal structure. This data-driven trend is spreading across a wider range of materials systems, both in time and space. For example, the team led by Xie Jianxin and Su Yanjing from the University of Science and Technology Beijing explored the application of interpretable machine learning in materials science. They demonstrated that combining material knowledge with machine learning can significantly improve the model's generalization and predictive accuracy, opening up new perspectives for the development of materials science. The related research, titled "Interpretable Machine Learning Applications: A Promising Prospect of AI for Materials," was published in Advanced Functional Materials.
Paper address:
https://advanced.onlinelibrary.wiley.com/doi/abs/10.1002/adfm.202507734
A research team from Argonne National Laboratory in the United States has proposed a generative AI framework, GHP-MOFsassemble, which can randomly generate and assemble new MOF structures. This framework uses molecular dynamics simulations to screen for highly stable MOFs and uses a Crystal Graph Convolutional Neural Network (CGCNN) and Grand Canonical Monte Carlo simulations to test the MOFs' carbon dioxide adsorption capacity. The related research, titled "A generative artificial intelligence framework based on a molecular diffusion model for the design of metal-organic frameworks for carbon capture," was published in Communications Chemistry.
Paper address:
https://www.nature.com/articles/s42004-023-01090-2
A research team from the University of Oxford published a study titled "The amorphous state as a frontier in computational materials design," highlighting the critical role of machine learning in breaking through traditional limitations in materials design. The study demonstrated how recent advances in computational modeling and artificial intelligence can bridge the previously missing link between the atomic-scale structure, microscopic properties, and macroscopic functionality of amorphous solids.
Paper address:
https://www.nature.com/articles/s41578-024-00754-2
This series of studies together paints a clear picture: materials science is entering a new era of intelligence, and we are in the midst of a materials research transformation led by machine learning. More importantly, intelligence has gradually spread from the design and synthesis of new materials to application scenarios, which is bound to further promote the implementation of new materials.
References:
1.https://pubs.acs.org/doi/10.1021/cr300014x
Get high-quality papers and in-depth interpretation articles in the field of AI4S from 2023 to 2024 with one click⬇️









