Command Palette
Search for a command to run...
DeepMind Releases AlphaGenome, Which Predicts the Effects of Mutations on All Modalities and Cell Types in 1 Second

Google DeepMind's Alpha series adds a new member - AlphaGenome,It can more comprehensively and accurately predict how a single variation or mutation in a human DNA sequence affects a series of biological processes that regulate genes.
The AlphaGenome model takes a DNA sequence of up to 1 million base pairs as input and predicts thousands of molecular properties related to its regulatory activity.At the same time, the impact of gene variation or mutation can be evaluated by comparing the prediction results of variant and non-variant sequences. This model is built on the basis of DeepMind's previous genomic model Enformer, and complements the AlphaMissense model that focuses on the classification of protein coding region variations.
Jun Cheng, co-first author of the paper, said on his personal X account, "RNA splicing errors are a common cause of many diseases. For the first time, we have built a unified model that can simultaneously predict RNA-seq coverage, splicing sites, site usage, and the specific splicing junctions they form, thereby more comprehensively depicting the overall picture of splicing results." He also pointed out thatOne of AlphaGenome's important breakthroughs is "the ability to predict splicing junctions directly from sequences and use them to predict variant effects."
"This is a milestone in the field," said Caleb Lareau, MD, from Memorial Sloan Kettering Cancer Center.For the first time, we have a model that combines long context, single-base accuracy, and state-of-the-art performance.DeepMind has opened a preview version to non-commercial research users through the AlphaGenome API and plans to officially release the model in the future.
* Research paper link:
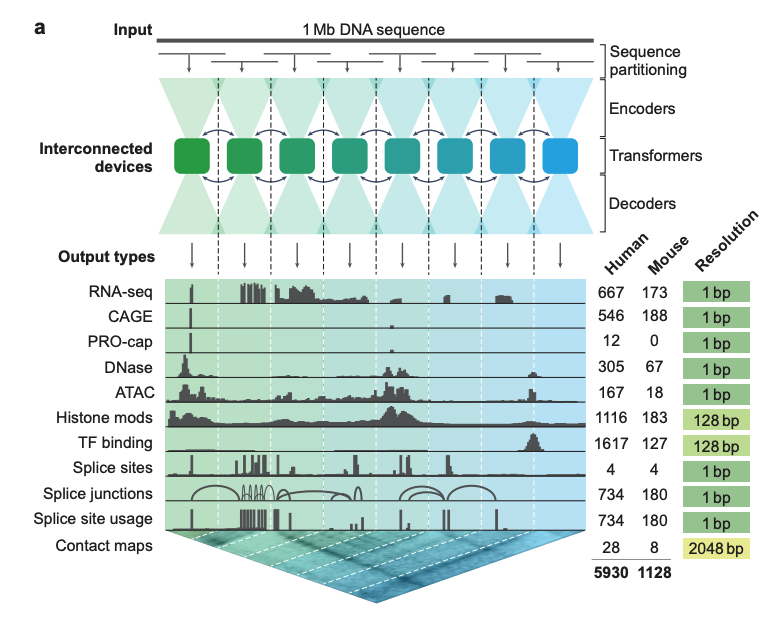
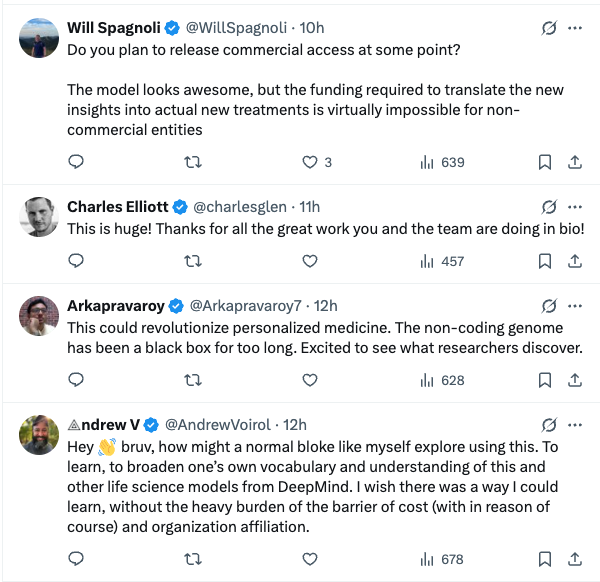
Based on 1 million DNA sequences and species information, using a U-Net-like design
As shown in Figure a below, the deep learning model AlphaGenome takes 1 Mb (million bases) of DNA sequence and species information (human/mouse) as input.5,930 human genome loci or 1,128 mouse genome loci for prediction of different cell typesCovers 11 output types, including:
* Gene expression (RNA-seq, CAGE, PRO-cap)
* Detailed splicing patterns (splice sites, splice site usage frequency, splice junctions)
* Chromatin state (DNase, ATAC-seq, histone modification, transcription factor binding)
* Chromatin contact map
In terms of model architecture,AlphaGenome adopts a U-Net-like backbone architecture design.As shown in Figure a below, the input sequence can be efficiently processed into two types of sequence representations:
1-dimensional embeddings (1 bp and 128 bp resolution): represent linear genomic sequences and are used to generate predictions of genomic trajectories.
* 2-dimensional embeddings (resolution 2048 bp): represent the spatial interactions between genomic fragments and are used to predict pairwise contact maps.
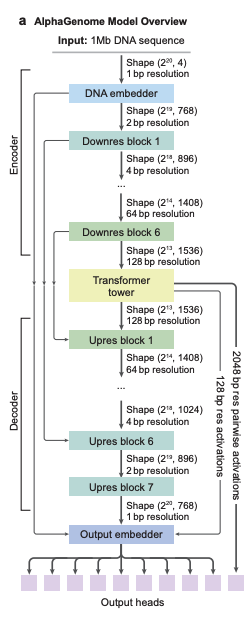
The convolutional layers of the model are used to model local sequence patterns to support fine-grained predictions, while the Transformer modules are used to model longer-range dependencies, such as interactions between enhancers and promoters. The model can be trained on a single base on a full 1Mb sequence, thanks to distributed sequence parallelism, and can run on 8 interconnected TPUv3 devices.
In terms of model training,The researchers adopted a two-stage training, namely pre-training and distillation.In the pre-training stage, it uses the existing experimental data to train two types of models, as shown in Figure b below:
* Fold-specific models:The training was performed using a four-fold cross-validation approach, i.e., using 3/4 of the segments in the reference genome for training and leaving the remaining 1/4 for validation and testing. These models were used to evaluate the generalization ability of AlphaGenome to predict genomic trajectories on unseen reference genome segments.
* All-folds models:The Teachers model is trained on all available segments of the reference genome as the next distillation stage, as shown in Figure c below.
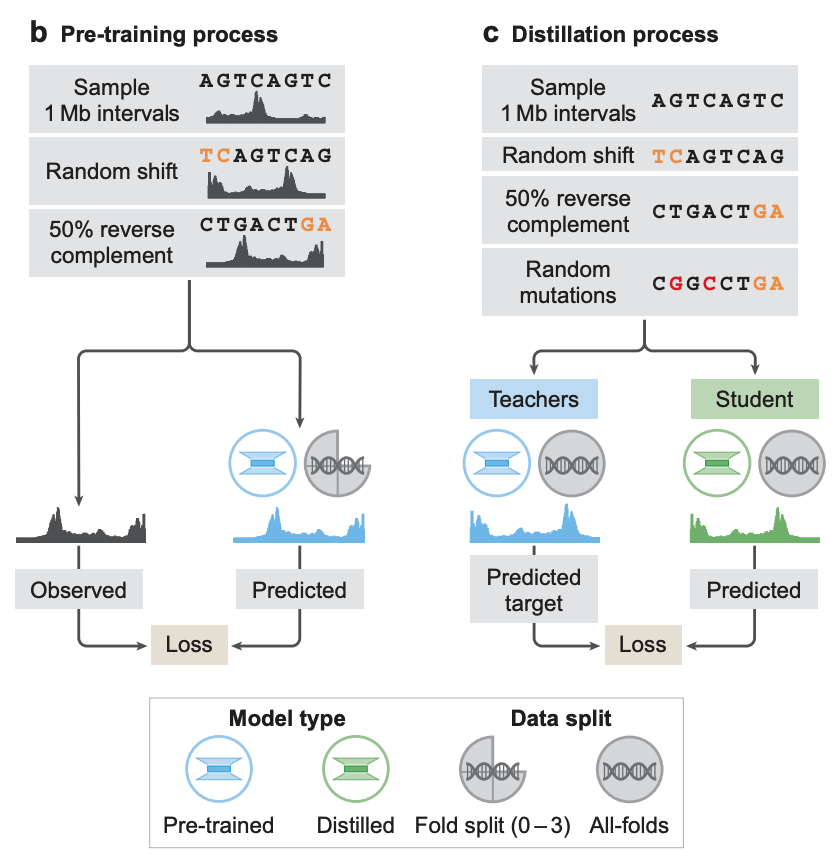
In the distillation phase, the researchers trained a Student model that shared a pre-trained architecture.The goal is to predict the combined output of multiple fully-folded teacher models using randomly augmented input sequences.Previous studies have shown that this distilled model can simultaneously achieve stronger robustness and higher variational effect prediction (VEP) accuracy in one model instance.
Thanks to this design, the Student model can complete the task of predicting the variation effects of all modalities and cell types with a single device call.On an NVIDIA H100 GPU, predictions for each variant take less than a second.This makes it extremely efficient in predicting large-scale variation effects compared to traditional multi-model integration methods.
AlphaGenome leads in various genome prediction tasks
According to DeepMind, AlphaGenome has the following unique advantages over existing methods:
Long sequence context + single base resolution
AlphaGenome can analyze DNA sequences up to a million bases long and make predictions at the single base level. This allows it to cover remote regions of regulatory genes while capturing fine biological details. Previous models often focused on balancing sequence length with prediction accuracy, limiting the range and accuracy of modalities that can be modeled. AlphaGenome's technological breakthrough breaks this limitation, with training using only half the computing resources of the original Enformer model and completing a training session in just 4 hours.
Comprehensive multimodal forecasting capabilities
The combination of high resolution and long input sequences enables AlphaGenome to predict unprecedentedly diverse regulatory patterns, providing researchers with more systematic gene regulation information.
Efficient mutation scoring
AlphaGenome can score the impact of variants within one second. By comparing the predicted differences in the sequences before and after the variant and using the most appropriate summary method for different modalities, it can quickly and accurately assess the potential impact of genetic variants on molecular mechanisms.
Novel splice site modeling
AlphaGenome innovatively enables the prediction of the location of RNA splicing junctions and their expression levels directly based on sequences. Many rare genetic diseases (such as spinal muscular atrophy and certain types of cystic fibrosis) are associated with splicing errors, and this capability provides a new tool for related etiology research.
Excellent performance in benchmarks
AlphaGenome leads in various genome prediction tasks, such as predicting DNA structural proximity, the effect of mutations on gene expression, and changes in splicing patterns. It beat the best existing models in 22 of the 24 DNA sequence prediction evaluations, and reached or exceeded the current best models in 24 of the 26 mutation effect tasks. More importantly, it is also the only model that can make joint predictions for all evaluation modalities, demonstrating strong versatility.
Specifically, to evaluate the performance of AlphaGenome’s models,The researchers first examined its generalization ability to unseen genomic segments, which is a prerequisite for achieving high-quality prediction of variant effects.They performed a total of 24 genomic trajectory prediction evaluations covering all 11 modalities predicted by the model. In cross-validation out-of-fold evaluations, the researchers used pre-trained fold-specific AlphaGenome models and compared their predictions with the current strongest external model in each task.
The results show thatAlphaGenome outperformed the corresponding external models in 22 of the 24 evaluations.As shown in Figure d below. It is worth noting that in the cell type-specific gene expression change (log-fold change, LFC) prediction task, AlphaGenome showed a relative performance improvement of +17.4% compared to another multimodal sequence model Borzoi, as shown in Figure e below.
In addition, AlphaGenome also surpasses specialized models that focus on a single modality in various tasks. For example:
In the prediction of chromatin contact map,AlphaGenome surpasses the Orca model, as shown by an increase of +6.3% in the Pearson correlation coefficient of the contact map and an increase of +42.3% in cell type-specific differences, as shown in Figure d below;
In the prediction of transcription start site tracks,AlphaGenome outperforms ProCapNet, with an overall count Pearson correlation coefficient improvement of +15%;
In chromatin accessibility prediction,AlphaGenome outperforms ChromBPNet, improving by +8% on ATAC-seq and +19% on DNase-seq.
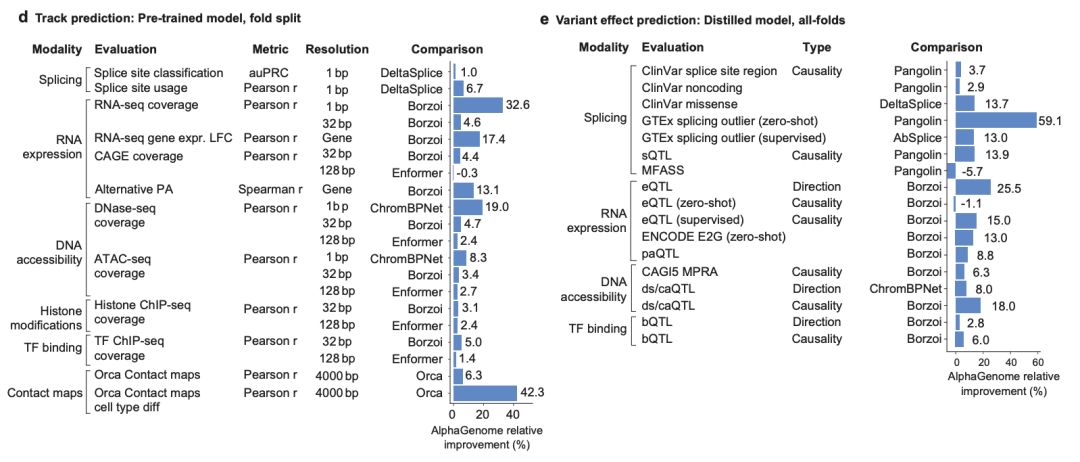
* Figure e: Relative performance improvement of AlphaGenome in the task of predicting partial variant effects.
Industry milestone highly praised
The launch of this blockbuster model by AlphaGenome has continued to spark heated discussions on Twitter since the news was released.
Pushmeet Kohli, vice president of research at DeepMind, said: “AlphaGenome provides a comprehensive view of the human non-coding genome by predicting the impact of DNA variants.It will deepen our understanding of disease biology and open up new avenues of research."In the comment section, in addition to exclamation and praise, everyone is more concerned about how to use it.
A PhD student in genetics from the University of Edinburgh said:"This model may completely redefine the way we discover disease-causing mutations and drug targets, which is of great significance."
A commentator in the field of biological sciences stated that "AlphaGenome is not just a single gene, but the entire regulatory genome. If DNA is compared to code, then AlphaGenome is the software composed of code."
In terms of practical applications, AlphaGenome has broad scientific research potential.For example, facingDisease mechanism research,It can more accurately predict the impact of genetic variation on regulatory processes, identify potential pathogenic variation, and reveal new targets. It is particularly suitable for studying rare variation with significant effects.existIn the field of synthetic biology,Direct the design of DNA for specific regulatory functions, such as activating a target gene only in nerve cells.existIn basic genomics research, it canAccelerate the localization and role definition of key functional elements, and help identify the "core instructions" required to regulate the function of specific cell types.
Professor Marc Mansour of University College London commented, "AlphaGenome provides key puzzle pieces for large-scale identification of the role of non-coding variants, allowing us to better understand complex diseases such as cancer." Currently, AlphaGenome is open to research for non-commercial purposes, and we look forward to more achievements from the academic community based on this.








