Command Palette
Search for a command to run...
Die Für ICML 2025 Ausgewählte Tsinghua-Universität/Renmin-Universität Von China/ByteDance Schlug Das Erste Molekülübergreifende Einheitliche Generierungsframework UniMoMo Vor, Um Das Design Mehrerer Arzneimittelmolekültypen Zu Erreichen

Die von Professor Liu Yang von der Tsinghua-Universität geleitete Gruppe, die von Professor Huang Wenbing von der Gaoling School of Artificial Intelligence an der Renmin-Universität China geleitete Gruppe und das Pharma-Team von ByteDance AI haben gemeinsam ein einheitliches Generationsframework für alle Molekülarten vorgeschlagen, UniMoMo.Dieses Framework stellt einheitlich verschiedene Arten von Molekülen basierend auf Molekülfragmenten (Blöcken) dar, verwendet Variational Autoencoder, um die vollständige Atomkonformation jedes Blocks zu komprimieren, und führt eine geometrische Diffusionsmodellierung im komprimierten latenten Raum durch, wodurch das Design verschiedener Arten von Bindungsmolekülen (kleine Moleküle, Peptide, Antikörper) für dasselbe Ziel erreicht wird. UniMoMo erzielt bei mehreren Benchmarks für molekulare Aufgaben Spitzenleistungen und demonstriert damit das große Potenzial des modalitätsübergreifenden Wissenstransfers und Datenaustauschs.

Die entsprechenden Ergebnisse wurden für ICML 2025 unter dem Titel „UniMoMo: Unified Generative Modeling of 3D Molecules for De Novo Binder Design“ ausgewählt.
Papieradresse:
Adresse des Open-Source-Projekts:
https://github.com/kxz18/UniMoMo
Warum Unified Modeling?
Verschiedene Molekültypen haben ihre eigenen Vor- und Nachteile bei der Arzneimittelentwicklung. Daher ist es oft notwendig, für verschiedene Krankheitsszenarien den am besten geeigneten Molekültyp auszuwählen. Zum Beispiel:
* Kleine Moleküle sind klein, leicht oral einzunehmen und haben eine starke Penetration, sodass sie in Zellen eindringen und auf Ziele einwirken können. Sie werden häufig bei chronischen Krankheiten und Stoffwechselerkrankungen eingesetzt.
* Peptidmoleküle haben hohe Zieleigenschaften und können an große und flache Bereiche auf der Oberfläche von Proteinen binden. Sie eignen sich für die gezielte Behandlung schwer medikamentös zu behandelnder Proteininteraktionsstellen und werden häufig bei der Behandlung von Krebs, Entzündungen usw. eingesetzt.
* Antikörper verfügen über eine extrem hohe Selektivität und Affinität und können bestimmte Proteinmarker stabil identifizieren, wodurch sie sich besonders für präzise Interventionsszenarien wie die Immuntherapie eignen.
Angesichts unterschiedlicher Krankheitsmechanismen, Zieleigenschaften und Medikamentenbedürfnisse eignen sich daher unterschiedliche Molekültypen. Bestehende generative Methoden modellieren normalerweise nur eine bestimmte Klasse von Molekülen (wie kleine Moleküle, Peptide oder Antikörper).Es kann weder den unterschiedlichen therapeutischen Anforderungen gerecht werden, noch die Gemeinsamkeiten zwischen verschiedenen Molekülen nutzen, um die Modellleistung zu verbessern.
Aus Anwendungssicht ermöglicht uns die einheitliche Modellierung, mehrere Arten von Arzneimittelkandidaten für dasselbe Ziel gleichzeitig zu untersuchen, wodurch mehr Optionen für verschiedene nachgelagerte Szenarien zur Verfügung stehen.
Aus der Perspektive des maschinellen Lernens weisen verschiedene Molekültypen ähnliche Bindungsregeln (Wasserstoffbrücken, π-π-Stapelung, Salzbrücken usw.) und geometrische Einschränkungen (Bindungslängen, Bindungswinkel usw.) auf und können voneinander lernen.Daher sollte eine einheitliche Modellierung in der Lage sein, die Generalisierungs- und plattformübergreifenden Übertragungsfähigkeiten des Modells durch die Nutzung eines größeren Datenumfangs zu verbessern.
Die Schwierigkeit der generativen einheitlichen Modellierung
Obwohl die Idee, verschiedene Arten von Molekülen gleichmäßig zu erzeugen, spannend ist, gibt es bei der Realisierung eines solchen Rahmens noch immer große Herausforderungen, vor allem bei der Wahl der Moleküldarstellung und der Gestaltung des Erzeugungsalgorithmus.
Erstens gibt es große Unterschiede in der Strukturdarstellung verschiedener Molekültypen: Kleine Moleküle bestehen aus verschiedenen funktionellen Gruppen und ihre Strukturen sind sehr unterschiedlich und nichtlinear; Während Peptide und Antikörper aus Aminosäuren bestehen, die in einer linearen Sequenz verbunden sind, weisen insbesondere Antikörper eine klare Unterteilung der funktionellen Regionen auf. Ein intuitiver, aber schlechter Ansatz besteht darin, alle Moleküle als Graphen von Atomen zu modellieren.Dieser Ansatz ignoriert jedoch die natürliche hierarchische Struktur von Molekülen, etwa wichtige Unterstrukturen wie Benzolringe oder Standardaminosäuren, und führt zu extrem hohen Rechenkosten, wenn es um Systeme mit großen Bindungsoberflächen wie Antikörpern geht.
Im Gegenteil, wenn nur gemeinsame strukturelle Fragmentvokabulare verwendet werden, um Graphen auf Fragmentebene zu konstruieren (z. B. berücksichtigt die meiste Proteindesignarbeit nur Cα koordinieren),Das Ignorieren von Details auf atomarer Ebene geht zu Lasten der Portabilität und Genauigkeit der Molekülerzeugung.Da die wesentlichen Gesetze beim Design von Bindungsmolekülen auf der räumlichen Interaktion mit dem Ziel und den geometrischen Beschränkungen innerhalb des Moleküls beruhen, handelt es sich hierbei um physikalische Gesetze, die auf atomarer Ebene definiert sind und eine präzise Unterstützung durch Informationen zu allen Atomen erfordern.
Um eine wirklich effektive und effiziente einheitliche molekulare Darstellung zu erstellen, müssen daher zwei Herausforderungen gleichzeitig gelöst werden:Es ist notwendig, die geometrischen Details auf atomarer Ebene beizubehalten und gleichzeitig die strukturellen hierarchischen Vorannahmen zu abstrahieren.
Zweitens: Wenn bei der Generierung Strukturfragmente eingeführt werden, um hierarchische Vorbedingungen zu bewahren, bringt dies grundlegende Herausforderungen für den Generierungsalgorithmus mit sich:Herkömmliche Diffusionsmodelle basieren normalerweise auf Datendarstellungen mit fester Länge und fester Struktur.Beispielsweise eine feste Anzahl von Punktwolken oder Atomen. Bei Strukturvorhersagemodellen wie AF3 führt der Diffusionsprozess nicht zu Änderungen der Atomzahl oder der 2D-Struktur, da die 2D-Topologie im Voraus gegeben ist. Für die Aufgabe der Molekülerzeugung müssen 2D-Topologie und 3D-Struktur gleichzeitig erzeugt werden. Wenn sich während des Entrauschungsprozesses die Art der Strukturfragmente ändert, ändern sich auch die entsprechende Anzahl, Art und Anordnung der Atome entsprechend. Dies widerspricht den Annahmen herkömmlicher Diffusionsmodelle und stellt extrem hohe Anforderungen an die Modellierung.
UniMoMo: Ein einheitliches generatives Modell
Um das Problem der großen strukturellen Unterschiede und der hohen Modellierungsschwierigkeiten verschiedener Molekültypen zu lösen, schlägt der Artikel ein neues Framework vor – UniMoMo.Es beginnt mit zwei Schlüsseldesigns, die die strukturelle Hierarchie und die Präzision auf atomarer Ebene effektiv berücksichtigen:
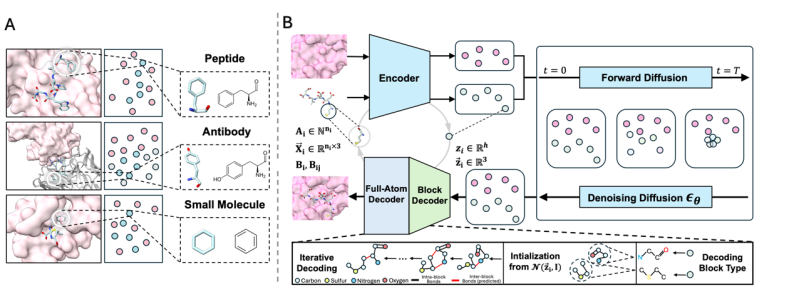
* Einheitliche Darstellung:Alle Molekültypen werden in Blockform modelliert.
Egal ob es sich um ein kleines Molekül, ein Peptid oder einen Antikörper handelt, UniMoMo stellt seine Struktur als Graph dar, der aus Molekülfragmenten (Blöcken) besteht. Jeder Block kann eine Standardaminosäure oder ein gewöhnliches kleines Molekülfragment (wie etwa ein Benzolring, Indol usw.) sein. Bei der Implementierung des Artikels umfassen die aufgezeichneten Molekülfragmente alle Standardaminosäuren und kleinen Molekülfragmente, die automatisch durch den Hauptalgorithmus für das Subgraph-Mining identifiziert werden. Alle unnatürlichen Aminosäuren können als kleine Moleküle klassifiziert werden.Diese Darstellung behält sowohl die Details der Moleküle auf atomarer Ebene als auch die hierarchische Struktur der verschiedenen Molekültypen selbst bei, wodurch eine einheitliche Modellierung möglich wird.
* Allatom-geometrisches implizites Raumdiffusionsmodell:Effiziente Generierung komprimierter Darstellungen.
Um das Problem der synchronen Änderungen der Art und Menge der Atome zu lösen, die durch Änderungen der Blocktypen während des Generierungsprozesses verursacht werden, und um die Generierungseffizienz und Strukturgenauigkeit zu verbessern,Der Artikel entwirft einen rein atomaren iterativen Variational Autoencoder (IterVAE).Alle Atome in jedem Block werden zu einem „Punkt“ im latenten Raum komprimiert, einschließlich eines Darstellungsvektors für den latenten Raum mit fester Länge und den entsprechenden Koordinaten des latenten Raums.
Das Modell führt dann in diesem komprimierten geometrischen latenten Raum eine generative Modellierung durch, um latente Darstellungen neuer Moleküle zu generieren, die schließlich wieder in die vollständige Atomstruktur dekodiert werden.Da die Datendarstellung des latenten Raums eine feste Länge hat (die Anzahl der Blöcke ist vorgegeben) und kontinuierlich ist, kann sie problemlos mit verschiedenen vorhandenen Generierungsalgorithmen kompatibel sein.In aktuellen Versuchen konnten Diffusionsmodelle relativ gute Ergebnisse liefern. Dieses Design ermöglicht es dem Modell, sich während des Generierungsprozesses auf das globale Layout zwischen den Blöcken zu konzentrieren, während die detaillierte Struktur auf atomarer Ebene vom Decoder vervollständigt wird, wodurch die Einheit aus hoher Effizienz und Genauigkeit auf atomarer Ebene erreicht wird.
Einheitliche Modellierung geht über die Modellierung einzelner Domänen hinaus
Um die Vielseitigkeit und Wirksamkeit von UniMoMo bei verschiedenen Molekültypen zu überprüfen, führten die Autoren eine systematische Bewertung in mehreren strukturbasierten Designaufgaben durch.Es umfasst drei repräsentative Typen von Bindungsmolekülen: kleine Moleküle, Peptide und Antikörper.Durch einen Vergleich mit dem repräsentativsten Modell zur Erzeugung einzelner Molekültypen im entsprechenden Bereich soll im Experiment untersucht werden, ob die einheitliche Modellierung über stärkere geometrische Modellierungsfähigkeiten und modalübergreifende Generalisierungsfähigkeiten verfügt, insbesondere im Hinblick auf Schlüsselindikatoren wie räumliche Strukturrationalität und Bindungsfähigkeit.
Die Ergebnisse zeigen, dassUniMoMo hat durch einheitliches Training eine umfassende Überlegenheit in allen Molekültypen erreicht.Es zeichnet sich nicht nur durch eine hohe Genauigkeit bei der Strukturwiederherstellung aus, sondern erzielt auch erhebliche Verbesserungen bei der geometrischen Rationalität der Schlüssel und der Qualität der Interaktion mit dem Ziel.
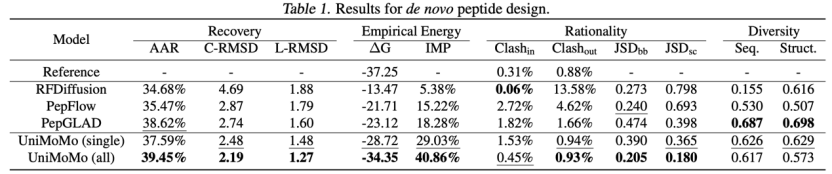
Bei der PeptidgenerierungsaufgabeUniMoMo übertrifft bestehende domänenspezifische Modelle bei mehreren Schlüsselindikatoren deutlich.Einschließlich RFDiffusion, PepFlow und PepGLAD usw. Insbesondere im Hinblick auf die strukturelle Genauigkeit erreichte UniMoMo einen niedrigeren RMSD von Komplexen und Monomeren, was darauf hindeutet, dass die von ihm erzeugten Peptidstrukturen näher an der tatsächlichen Bindungskonformation lagen.
UniMoMo kann auch Strukturen mit niedrigeren Rosetta-Bindungsenergien erzeugen.Dies spiegelt seine stärkere Modellierungsfähigkeit für die geometrischen Merkmale von Proteinbindungsstellen wider.Darüber hinaus zeigte UniMoMo auch eine führende Leistung bei Indikatoren der geometrischen Rationalität, wie etwa der Konsistenz der Diederwinkelverteilung (JSD von Rückgrat-/Seitenkettentorsionen) und räumlichen Konflikten auf atomarer Ebene (Kollisionsrate), die die Qualität der Peptidkonformation messen. Darüber hinaus übertraf UniMoMo (alle), das mit allen Daten trainiert wurde, bei verschiedenen Indikatoren durchweg das Modell, das nur mit Peptiddaten trainiert wurde.Die Fähigkeit von UniMoMo, über molekulare Arten hinweg zu lernen und zu verallgemeinern, wird demonstriert.
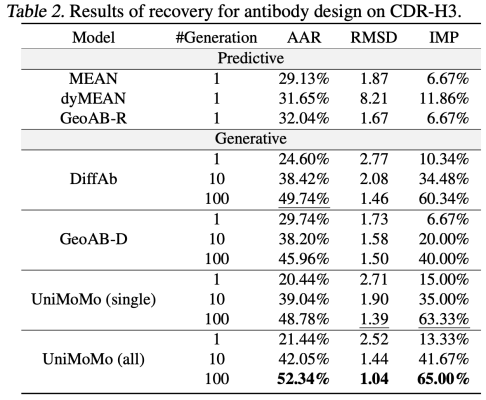
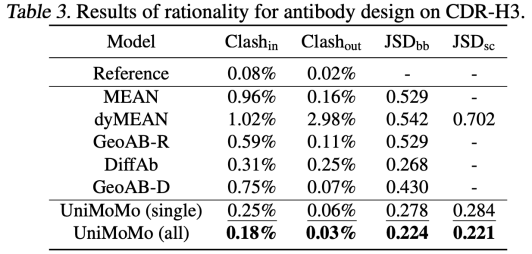
UniMoMo zeigte auch bei der Aufgabe des Antikörperdesigns eine starke Leistung. Im Vergleich zu bestehenden Methoden wie MEAN, dyMEAN und DiffAb,UniMoMo übertrifft alle anderen Ziele hinsichtlich wichtiger Indikatoren wie dem Abruf natürlich gebundener Sequenzen und Strukturen (AAR und RMSD) und der Verbesserung der Bindungsenergie (IMP).Insbesondere bei der Auswertung der Generierung mehrerer Proben ist UniMoMo in der Lage, mit höherer Wahrscheinlichkeit Antikörperfragmente zu erzeugen, die der natürlichen Konformation nahe kommen, und zeigt damit seine guten Explorationsfähigkeiten im Bereich der Antikörperstrukturen.
Ebenso übertrifft UniMoMo(all), das gemeinsam mit Daten verschiedener Molekültypen trainiert wird, die Version, die nur mit Antikörperdaten trainiert wurde, in allen Indikatoren.Dies zeigt, dass die einheitliche Modellierung dem Modell dabei hilft, universellere und übertragbarere räumliche Gesetze molekularer Strukturen zu erlernen.Dieses Ergebnis unterstreicht die Gemeinsamkeiten bei der Strukturmodellierung zwischen verschiedenen Molekültypen und bestätigt den erheblichen Wert der domänenübergreifenden Datenfusion für die Verbesserung der Generierungsqualität.
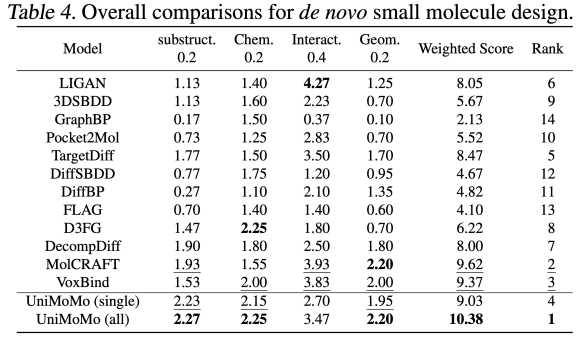
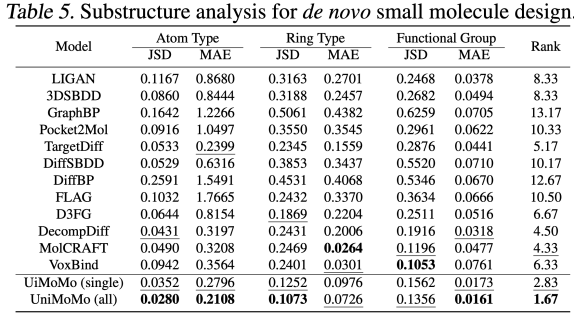
UniMoMo zeigte auch bei der Erzeugung kleiner Moleküle eine überragende Leistung. Durch die Auswertung des CrossDocked2020-DatensatzesDie Autoren stellten fest, dass UniMoMo bei der umfassenden Evaluierung auf Basis von CBGBench bestehende Mainstream-Methoden übertraf.
Insbesondere erzielte UniMoMo höhere Gesamtpunktzahlen in Bezug auf die Unterstrukturverteilung (Atomtypen, funktionelle Gruppen usw.), die Rationalität chemischer Eigenschaften (QED, LogP, SA usw.), die Qualität der geometrischen Struktur (Bindungslängen-/Winkelverteilung und atomare Konfliktrate usw.) und die Interaktionspunktzahl (Vina-Docking) (die vollständigen experimentellen Ergebnisse finden Sie im Originaltext). Wichtig ist, dass UniMoMo(all), das über verschiedene Molekültypen hinweg trainiert wird, in allen Bewertungsdimensionen signifikante Verbesserungen im Vergleich zur Single-Domain-Version zeigt, die nur mit Daten kleiner Moleküle trainiert wurde. Dies zeigt, dassSogar im Szenario kleiner Moleküle mit der flexibelsten Molekülstruktur und den unterschiedlichsten Typen kann das einheitliche Modell immer noch geometrische Gesetze und Interaktionsmuster von anderen Molekültypen übertragen und dadurch die Rationalität der Monomerkonformation und der relativen Taschenraumaufteilung des kleinen Moleküls verbessern.Dieses Phänomen bestätigt einmal mehr das Kernkonzept von UniMoMo: Die geometrischen Beschränkungen und Bindungsmechanismen zwischen verschiedenen Molekülen weisen gemeinsam nutzbare Muster auf, und eine einheitliche Modellierung kann dieses Potenzial effektiv stimulieren.
Durch die Kombination der experimentellen Ergebnisse der drei Aufgabentypen zeigt UniMoMo äußerst konsistente Vorteile: Das vereinheitlichte Modell, das mit molekularen Speziesdaten trainiert wurde, übertrifft das vorhandene Single-Domain-Generation-Modell bei seinen jeweiligen Aufgaben und verfügt im Vergleich zu UniMoMo, das nur mit Single-Domain-Daten trainiert wurde, über deutlich verbesserte Fähigkeiten. Dieses Phänomen zeigt, dass die scheinbar unterschiedlichen Aufgaben im Moleküldesign tatsächlich ein hohes Maß an Gemeinsamkeiten in den zugrunde liegenden physikalischen und chemischen Einschränkungen und räumlich-geometrischen Gesetzen aufweisen.Die einheitliche Modellierungsstrategie von UniMoMo erfasst und verstärkt diese Gemeinsamkeit und erreicht dadurch eine aufgabenübergreifende Übertragung und ergänzende Verbesserung.Diese Ergebnisse bestätigen nicht nur die Wirksamkeit von UniMoMo, sondern liefern auch starke empirische Unterstützung für den Aufbau eines leistungsfähigeren einheitlichen Molekülgenerierungssystems in der Zukunft.
GPCR-Fallstudien
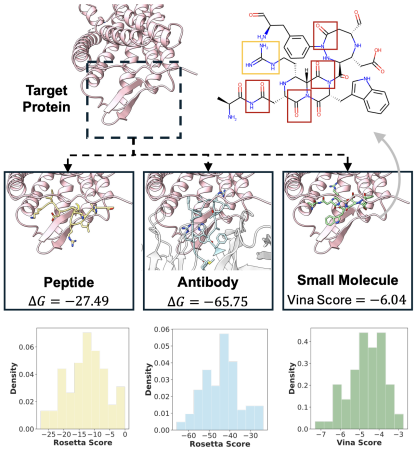
Als Fallstudie wählten die Autoren eines der wichtigsten Arzneimittelziele beim Menschen, den G-Protein-gekoppelten Rezeptor (GPCR), um die Fähigkeit von UniMoMo zu bewerten, verschiedene Arten von Molekülen (Peptide, Antikörper, kleine Moleküle) an derselben Bindungsstelle zu erzeugen. Die von UniMoMo erzeugten Peptide, Antikörper und kleinen Moleküle zeigen alle eine gute Verteilung unter den Kraftfeldern, die üblicherweise zur Bewertung der Bindungsenergie verwendet werden (Rosetta ΔG, Vina-Score).Noch überraschender ist, dass die generierte kleine Molekülstruktur auch spontan funktionelle Gruppen simuliert, die natürlichen Aminosäureseitenketten ähneln und zum Aufbau von Wasserstoffbrücken und zur Bildung wichtiger Wechselwirkungen mit dem Ziel verwendet werden. Darüber hinaus übernehmen kleine Moleküle auch lokale geometrische Konfigurationen von Peptiden und Antikörpern, wie etwa Amidverbindungen am Molekülrückgrat, die es ihnen ermöglichen, Bindungstaschen effektiv zu füllen, die ursprünglich eher für große Moleküle geeignet sind. Dieser Fall demonstriert anschaulich die Fähigkeit von UniMoMo, über verschiedene Modalitäten hinweg zu leihen und sich automatisch an Bindungstaschen in tatsächlichen Aufgaben anzupassen, und spiegelt sein Potenzial wider, die Interaktion zwischen Zielen und Molekülen und die internen geometrischen Beschränkungen von Molekülen auf der dreidimensionalen Strukturebene gründlich zu verstehen.
Zukünftige Erkundung
Obwohl UniMoMo starke Fähigkeiten zur einheitlichen Generierung in mehreren Molekültypen und Aufgaben gezeigt hat, wiesen die Autoren auch darauf hin, dass es in dieser Richtung noch viele zukünftige Möglichkeiten gibt, die es wert sind, erforscht zu werden.
Die aktuelle Arbeit konzentriert sich hauptsächlich auf die Modellierung natürlicher Aminosäuren und häufiger Molekülfragmente, die auf komplexere Arzneimittelformen wie nicht-natürliche Aminosäuren, nachträglich modifizierte Peptide/Antikörper, zyklische Moleküle usw. ausgeweitet werden kann, wodurch ein breiteres Spektrum an Kandidatenmolekülen abgedeckt wird. Das Konzept der einheitlichen Modellierung bietet außerdem die Möglichkeit, die Steuerbarkeit und Interpretierbarkeit des Modells zu untersuchen, und dürfte die Entwicklung generativer Modelle zu zuverlässigeren und praktischeren Plattformen für das molekulare Design weiter vorantreiben. Zusammenfassend lässt sich sagen, dass die Einführung von UniMoMo nicht nur einen allgemeinen und leistungsstarken generativen Rahmen für molekulare Designaufgaben bietet, sondern auch eine neue Richtung voller Potenzial für die KI-gesteuerte Arzneimittelforschung eröffnet.








