Command Palette
Search for a command to run...
Ausgewählt Für NeurIPS 2024! Die Westlake University Hat Das Universelle Molekulare Umkehrfaltungsmodell UniIF Vorgeschlagen, Das AlphaFold 3 Weiter ergänzt.

Die inverse molekulare Faltung spielt eine Schlüsselrolle bei der Entwicklung von Medikamenten und Materialien, da sie es Wissenschaftlern ermöglicht, neue Moleküle mit gewünschten Strukturen zu synthetisieren. Frühere Studien konzentrierten sich meist auf die umgekehrte Faltung von Makromolekülen oder kleinen Molekülen, selten jedoch auf die umgekehrte Faltung allgemeiner Moleküle.
Bei der Erstellung eines einheitlichen Gesamtmodells gibt es drei große Herausforderungen:① Einheitenunterschiede: Große Moleküle verwenden im Allgemeinen vordefinierte Mikrostrukturen als Grundeinheiten, wie etwa Aminosäuren für Proteine und Nukleotide für RNA; kleine Moleküle verwenden Atome als Grundeinheiten; 2. Extraktion geometrischer Merkmale: Verschiedene Studien verwenden unterschiedliche Strategien zur Extraktion geometrischer Merkmale, wie z. B. Entfernung, Winkel und Tensorprodukt, und es fehlt eine einheitliche Charakterisierungsmethode. ③ Systemskala: Kleine Moleküle ermöglichen es globalen Aufmerksamkeitsmechanismen, langfristige Abhängigkeiten zu erlernen, was bei großen Molekülen jedoch oft nicht funktioniert.
Um die oben genannten Herausforderungen zu bewältigen und die Fortschritte von RoseTTAFold All-Atom und AlphaFold 3 bei der Vorhersage molekularer Strukturen weiter zu ergänzen,Ein Team des Future Industry Research Center der Westlake University schlug ein einheitliches Modell, UniIF, für die inverse Faltung aller Moleküle vor.Die Forscher führten umfassende Experimente zu mehreren Aufgaben durch, darunter Proteindesign, RNA-Design und Materialdesign, um die Wirksamkeit von UniIF zu demonstrieren. Die Ergebnisse zeigen, dass UniIF bei allen Aufgaben eine hochmoderne Leistung erzielt.
Die zugehörige Forschung mit dem Titel „UniIF: Unified Molecule Inverse Folding“ wurde für die Top-Konferenz NeurIPS 2024 ausgewählt.
Forschungshighlights:
* Das vorgeschlagene einheitliche Modell UniIF bietet eine vielseitige und effektive Lösung für die allgemeine molekulare inverse Faltung
* Das Modell ist auf zwei Ebenen vereinheitlicht: Auf der Datenebene wird eine einheitliche Blockdiagramm-Datenform für alle Moleküle vorgeschlagen, einschließlich der Konstruktion des lokalen Koordinatensystems und der Initialisierung geometrischer Merkmale; Auf Modellebene wird ein geometrisches Block-Attention-Netzwerk eingeführt, um die dreidimensionalen Interaktionen aller Moleküle zu erfassen
* Die Forscher zeigten, dass die vorgeschlagene Methode den modernsten Methoden in drei Hauptaufgaben überlegen ist: Proteindesign, RNA-Design und Materialdesign. Diese Errungenschaft könnte sich positiv auf die Bereiche maschinelles Lernen, Arzneimittelforschung und Materialwissenschaften auswirken.
Papieradresse:
https://arxiv.org/abs/2405.18968
Folgen Sie dem offiziellen Konto und antworten Sie mit „Molecular Reverse Folding“, um das vollständige PDF zu erhalten
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Wählen Sie den entsprechenden Datensatz für drei Aufgabenexperimente aus
Bei der ProteindesignaufgabeDie Forscher evaluierten UniIF anhand des CATH4.3-Datensatzes. Der Datensatz wird gemäß dem topologischen Klassifizierungscode von CATH aufgeteilt, was zu 16.631 Trainingsbeispielen, 1.516 Validierungsbeispielen und 1.864 Testbeispielen führt.
Um die Generalisierungsfähigkeit zu bewerten, haben die Forscher eine Zeitpartitionierungsstrategie angewendet, da einige Baselines vorab trainierte ESM2-Modelle verwenden, was ein Risiko von Datenlecks birgt. Bei der zeitpartitionierten Auswertung werden Daten vor einem bestimmten Datum dem Trainingsdatensatz und Daten nach diesem Datum dem Testdatensatz zugeordnet. Für die zeitliche Auswertung der Strukturen wird der CASP15-Datensatz verwendet, der neue Kristallstrukturen enthält, die während des Trainings nicht gesehen wurden. Für die zeitliche Auswertung der Sequenzen wird der NovelPro-Datensatz verwendet, der 76 Proteinsequenzen enthält, die innerhalb von 30 Tagen vor dem 23. November 2023 veröffentlicht wurden, und deren Strukturen von AlphaFold 2 vorhergesagt werden.
Bei RNA-DesignaufgabenDie Forscher führten RNA-Experimente an einem von RDesign gesammelten Datensatz durch, der 2.218 RNA-Tertiärstrukturen enthält, die basierend auf ihrer strukturellen Ähnlichkeit in einen Trainingssatz (1.774 Strukturen), einen Testsatz (223 Strukturen) und einen Validierungssatz (221 Strukturen) unterteilt sind. Aufgrund der geringen Anzahl an Datenproben berichteten die Forscher über die mittlere Wiederfindungsrate und ihre Standardabweichung aus drei unabhängigen Durchläufen.
In der MaterialdesignaufgabeDie Forscher werteten UniIF anhand des CHILI-3K-Datensatzes aus, der aus Bildern von Nanomaterialien besteht, die aus einzelnen Metalloxiden gewonnen wurden. Der Datensatz umfasst 53 Metallelemente und ein Nichtmetallelement (Sauerstoff) mit insgesamt 3.180 Graphen, 6.959.085 Knoten und 49.624.440 Kanten.
Modellarchitektur: UniIF, ein einheitliches Modell für allgemeine molekulare inverse Faltung
Wie in der Abbildung unten gezeigt, schlugen die Forscher ein einheitliches Modell für die allgemeine inverse molekulare Faltung vor.
① Das Modell wandelt alle Arten von Molekülen in Blockdiagramme um – für Makromoleküle wird ein vordefiniertes Framework basierend auf Aminosäuren und Nukleotiden verwendet; für kleine Moleküle wird durch eine GNN-Schicht ein lokales Framework für jeden Block gelernt;
2. Verwenden Sie den Geometric Featurizer, um die geometrischen Knoten- und Kantenfunktionen zu initialisieren.
3. Es wird eine Blockgraph-Aufmerksamkeitsschicht vorgeschlagen, auf deren Grundlage ein Blockgraph-Neuralnetzwerk aufgebaut wird, um zu lernen, reichhaltige Blockdarstellungen auszudrücken.
④ Schließlich zeigen wir, dass UniIF bei einer Vielzahl von Aufgaben, darunter Proteindesign, RNA-Design und Materialdesign, wettbewerbsfähige Ergebnisse erzielen kann.
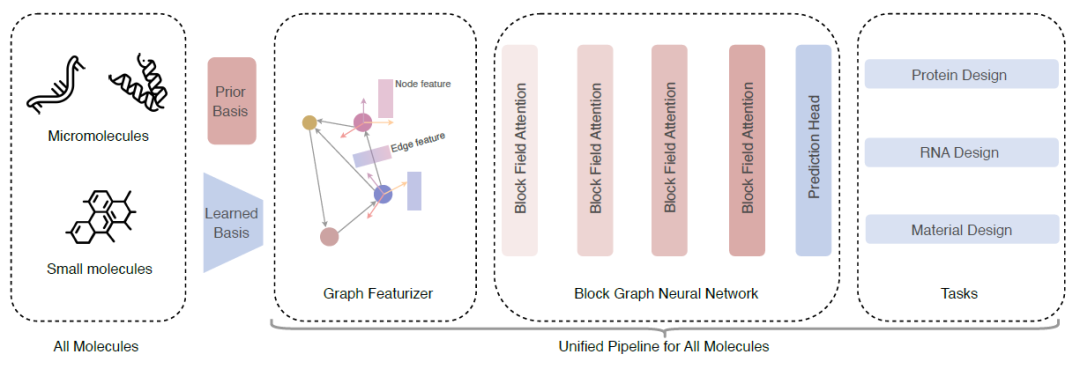
Bausteindiagramm:Der erste Schritt der Modellarchitektur besteht darin, Blockdiagramme zur Darstellung aller Arten von Molekülen einzuführen. Der Schlüssel liegt darin, unregelmäßige Atomsätze (unterschiedlicher Größe) in reguläre Blockdarstellungen (fester Größe) umzuwandeln. Um die Modellierung aller Moleküle zu vereinheitlichen, führten die Forscher eine rahmenbasierte Blockdarstellung ein, wobei ein Block einen äquivarianten Rahmen und invariante Eigenvektoren enthält und ein lokaler Rahmen eine Achsenmatrix und einen Verschiebungsvektor enthält. Bei großen Molekülen ist die Achsenmatrix auf Basis von Aminosäuren und Nukleotiden vordefiniert; Da es für kleine Moleküle kein a priori gemeinsames Strukturmuster gibt, muss die Achsenmatrix erlernt werden. Bei einem aus n Blöcken bestehenden Molekül verwendeten die Forscher den kNN-Algorithmus, um einen Blockgraphen zu erstellen.
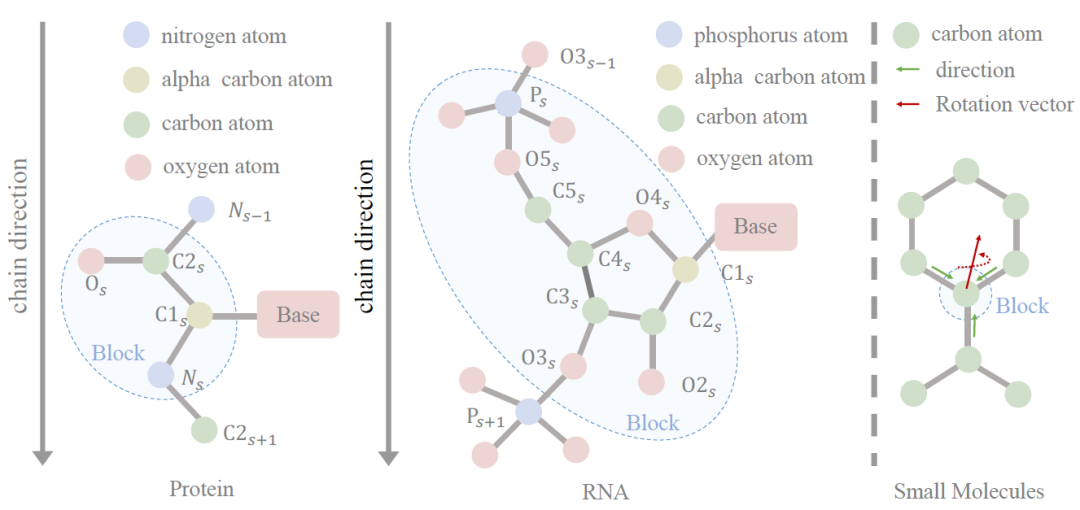
Extraktion von Blockdiagramm-Funktionen:Für kleine Moleküle sind keine vordefinierten lokalen Rahmen verfügbar, daher müssen Forscher für jedes Atom einen lokalen Rahmen lernen. Das heißt, bei einem gegebenen Molekül verwenden sie eine GNN-Schicht, um die atomare Darstellung zu initialisieren, und verwenden dann einen geometrischen Merkmalsextraktor, um die geometrischen Knoten- und Kantenmerkmale zu initialisieren.
Blockgraph-Aufmerksamkeitsmodul:Die Forscher führten ein geometrisches Blockaufmerksamkeitsnetzwerk ein, das geometrische Interaktions-, Interaktionsaufmerksamkeits- und virtuelle Langzeitabhängigkeitsmodule umfasst, um die dreidimensionalen Interaktionen aller Moleküle zu erfassen.
Forschungsergebnisse: UniIF übertrifft modernste Methoden bei allen Aufgaben
Die Forscher demonstrierten die Wirksamkeit von UniIF durch mehrere inverse Faltungsaufgaben und Ablationsstudien, darunter:
* Proteindesign (T1): Entwurf von Proteinsequenzen, die sich in Zielstrukturen falten können
* RNA-Design (T2): Entwerfen Sie RNA-Sequenzen, die sich in Zielstrukturen falten können
* Materialdesign (T3): Entdeckung stabiler Zusammensetzungen aus bekannten Materialstrukturen
① Proteindesign (T1)
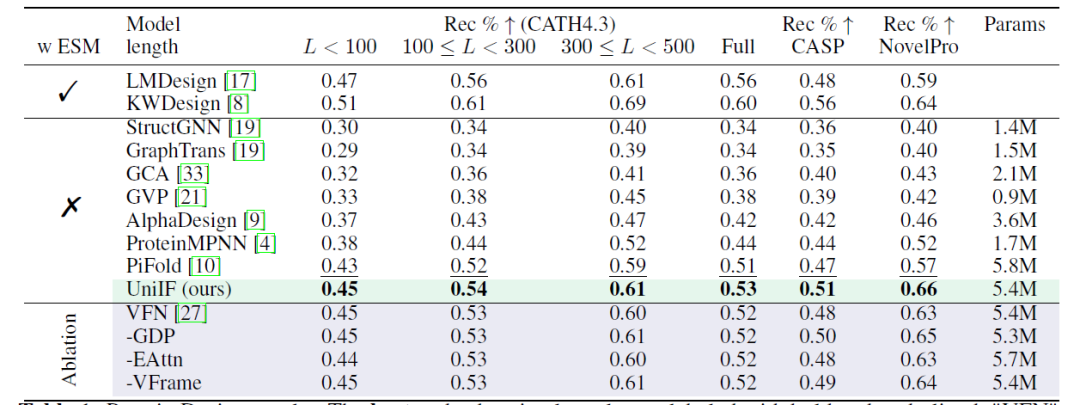
Ziel des Proteindesigns ist die Entwicklung von Proteinsequenzen, die sich in eine Zielstruktur falten können. Die Forscher liefern Ergebnisse unter verschiedenen Bedingungen (mit und ohne ESM2) und in mehreren Datensätzen (CATH4.3, CASP, NovelPro). Wie in der folgenden Tabelle gezeigt: Mit einem reinen inversen Faltungsmodell ohne ESM2 erzielt UniIF die beste Leistung bei allen Datensätzen und demonstriert damit seine Wirksamkeit.
*LMDesign und KWDesign enthalten ESM2; StructGNN, GraphTrans, GCA, GVP, AlphaDesign, ProteinMPNN und PiFold enthalten kein ESM2
Bei CATH4.3 ist die allgemeine Verbesserung aufgrund des starken Basismodells begrenzt, aber die zeitaufgeteilte Auswertung unterstreicht den Vorteil von UniIF hinsichtlich der Generalisierungsfähigkeit. UniIF übertrifft die starke Basislinie PiFold mit weniger lernbaren Parametern. Bei der Bewertung der Zeitpartitionierung übertrifft UniIF alle Basislinien, einschließlich des auf ESM2 basierenden Ansatzes, bei weitem. Bei NovelPro mit neuen Sequenzen übertraf UniIF bei der Verwendung von ESM2 zur Sequenzoptimierung LMDesign und KWDesign.Dies weist darauf hin, dass UniIF über eine überlegene Generalisierungsfähigkeit verfügt, die für praktische Anwendungen von entscheidender Bedeutung ist.
② RNA-Design (T2)
Das Ziel des RNA-Designs besteht darin, RNA-Sequenzen zu entwerfen, die sich in eine Zielstruktur falten können. Wie aus der folgenden Tabelle hervorgeht, erzielt UniIF in allen Fällen die beste Leistung. Dies stellt eine erhebliche Verbesserung dar, da zuvor starke Basismodelle wie PiFold nur im Proteindesign herausragend waren. Es wird berichtet, dassUniIF ist das erste Modell, das sowohl bei Protein- als auch bei RNA-Designaufgaben Spitzenleistung erzielt und damit seine Vielseitigkeit und Effektivität unter Beweis stellt.
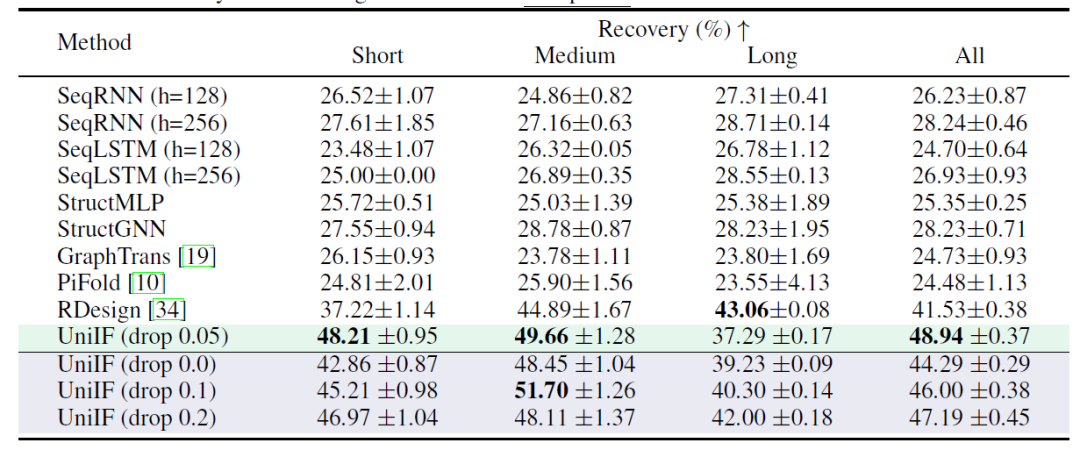
3 Materialdesign (T3)
Das Auffinden stabiler Atomkombinationen innerhalb bekannter Materialstrukturen ist für die Entdeckung neuer Materialien von entscheidender Bedeutung. Daher bewerteten die Forscher auch die Leistung von UniIF bei dieser neuen Aufgabe. Wie in der folgenden Tabelle gezeigt,UniIF übertrifft alle Basismodelle deutlich.
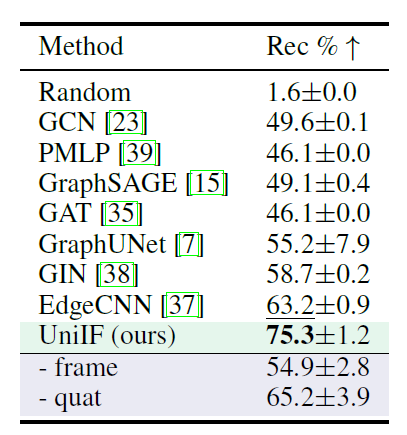
④ Fallstudie
In der folgenden Abbildung zeigen die Forscher die entworfenen Protein- und RNA-Sequenzen. Darüber hinaus wurde die entworfene Sequenz mithilfe von AlphaFold 3 in die Struktur zurückgefaltet – die wahre Struktur (grau), die PiFold-Struktur (grün) und die UniIF-Struktur (pink) wurden ausgerichtet und verglichen. Die Forscher stellten fest, dassUniIF erzielt Verbesserungen sowohl bei der Wiederherstellungsrate als auch bei der mittleren quadratischen Abweichung (RMSD) und beweist damit seine Wirksamkeit bei der inversen Faltungsaufgabe.

UniIF-Modell ergänzt AlphaFold 3 weiter
Das allgemeine molekulare Lernen hat in den letzten Jahren zunehmend an Aufmerksamkeit gewonnen und RoseTTAFold All-Atom (RFAA) und AlphaFold 3 sind zwei repräsentative Modelle, die in dieser Richtung bemerkenswerte Erfolge erzielt haben.
Am 7. März 2024 veröffentlichte David Baker in Science eine Forschungsarbeit mit dem Titel „Generalized biomolecular modeling and design with RoseTTAFold All-Atom“. Das Team entwickelte RoseTTAFold All-Atom (RFAA), das rückstandsbasierte Darstellungen von Aminosäuren und DNA-Basen mit atomaren Darstellungen aller anderen Gruppen kombiniert, um Baugruppen aus Proteinen, Nukleinsäuren, kleinen Molekülen, Metallen und kovalenten Modifikationen einer gegebenen Sequenz und chemischen Struktur zu modellieren.
Originalarbeit:
https://www.science.org/doi/10.1126/science.adl2528
Am 9. Mai 2024 veröffentlichten Demis Hassabis, John Jumpe und andere in Nature eine Forschungsarbeit mit dem Titel „Accurate structure prediction of biomolecular interactions with AlphaFold 3“. Die Studie brachte AlphaFold 3 auf den Markt, ein neues Modell, das die Struktur von Komplexen vorhersagen kann, die fast alle Molekültypen in der Proteindatenbank enthalten, einschließlich der Art und Weise, wie sich Liganden (kleine Moleküle), Proteine und Nukleinsäuren (DNA und RNA) zusammenfügen und miteinander interagieren. Darüber hinaus kann das Modell die strukturellen Auswirkungen posttranslationaler Modifikationen und Ionen auf diese Molekülsysteme vorhersagen und so Forschern dabei helfen, die Struktur biologischer Molekülsysteme auf atomarer Ebene genau zu beobachten.
Originalarbeit:
https://www.nature.com/articles/s41586-024-07487-w
Bei genauerer Betrachtung der beiden Modelle verwendet RFAA Atombindungsdiagramme zur Darstellung kleiner Moleküle und Gerüstdiagramme zur Darstellung großer Moleküle. AlphaFold 3 verwendet eine zweischichtige Darstellung, nämlich Atomdarstellung und Labeldarstellung, die auf alle Moleküle anwendbar ist. Das Tag-Konzept entspricht dem oben erwähnten Blockkonzept und stellt eine Gruppe von Atomen dar, beispielsweise Aminosäuren oder Nukleotide.
GET und EPT sind zwei kürzlich vorgeschlagene Modelle, die eine Blockdarstellung sowohl für kleine als auch für große Moleküle übernehmen und neue isotrope Transformator-Frameworks einführen. Im Gegensatz zu RFAA, das Atombindungsdiagramme für kleine Moleküle spezifiziert, verwendet das in diesem Artikel vorgestellte UniIF-Modell ein einheitliches Blockdiagramm für alle Molekültypen, für das keine Atombindungsdiagramme erforderlich sind. Darüber hinaus führt das Modell auch eine Vektorbasis für jeden Block ein, die sich von AlphaFold 3, GET und EPT unterscheidet.
Da die Herausforderung, ein universelles Molekülmodell zu entwickeln, teilweise gelöst ist,Das UniIF-Modell kann als weitere Ergänzung der Fortschritte bei der Vorhersage molekularer Strukturen durch „Vorgänger“ wie RoseTTAFold All-Atom und AlphaFold 3 angesehen werden.In Zukunft wird die kontinuierliche Iteration großer biologischer Modelle den Forschern dabei helfen, die biologische Welt neu zu verstehen und die Arzneimittelforschung neu zu überdenken, was der gesamten Menschheit zugutekommt.
Quellen:
1.https://arxiv.org/abs/2405.18968
2.https://mp.weixin.qq.com/s/8OvxVlUuZZZ2gcepIl5UBw
3.https://www.jiqizhixin.com/articles/2024-03-08-6
4.https://m.thepaper.cn/newsDetail_forward_28984037









