Command Palette
Search for a command to run...
推理速度提升 3 倍,多伦多大学等提出 dnaHNet,基因组学习计算成本降低近 4 倍

基因组是生物体全部遗传信息的载体,决定着细胞的功能、个体的发育以及物种的演化方向。隐藏在序列之中的「DNA 语法」,构成了生命运行的底层规则,也是现代生物学亟待破解的核心问题之一。对这一语法的理解,不仅关乎基础科学认知,更直接影响疾病诊断、药物研发以及合成生物学等关键应用的发展。
近年来,基于大规模序列数据预训练的基础模型逐渐成为破解这一问题的重要路径。随着算力、数据规模与模型参数的持续提升,这类模型展现出类似「缩放定律」的性能增长趋势。以 Nucleotide Transformer 、 Evo 为代表的模型,参数规模已扩展至数十亿级,并在跨物种序列上完成训练,在变异效应预测、调控元件解析等任务中取得了显著进展。
然而,DNA 序列本质上是一条连续且缺乏明确边界的核苷酸链,这一点与自然语言存在根本差异。当前主流的两类建模范式——固定分词与单核苷酸级建模——分别在表达能力与计算效率之间存在明显权衡:前者可能破坏生物学功能单元,后者则带来高昂的计算成本。因此,如何在计算可行性与生物学保真度之间取得更优平衡,成为关键挑战。动态分词作为潜在解决方案,仍有待系统性探索。
在这一背景下,多伦多大学、加拿大 Vector 人工智能研究院及美国 Arc Institute 等机构联合提出的 dnaHNet 模型,为突破上述瓶颈提供了新的思路。相关研究成果以「dnaHNet: A Scalable and Hierarchical Foundation Model for Genomic Sequence Learning」为题,已发表预印本于 arXiv 。
研究亮点
* dnaHNet 计算效率超越 StripedHyena2,推理速度较 Transformer 提升 3 倍以上。
* 提出压缩率调度、编码器-解码器平衡等最优训练策略。
* 在变异效应预测与基因必需性分类等零样本任务上达前沿水平。
* 可学习上下文相关的生物学分词,适配密码子、启动子、基因间区等功能区域。

论文地址:
https://arxiv.org/abs/2602.10603
关注公众号,后台回复「dnaHNet」获取完整 PDF
用于模型训练与评估的多层次基因组数据集设计
为支撑模型训练与系统评估,该研究构建了多层次的数据体系。预训练数据来源于基因组分类数据库(GTDB)处理后的子集,并严格遵循 Evo 模型 OpenGenome 数据集的过滤、质控与去冗余流程。筛选标准涵盖组装完整性、污染水平及标记基因含量等关键指标;在通过筛选后,每个物种级聚类仅保留一个代表性基因组。
最终数据集覆盖 85,205 种原核生物,包含 17,648,721 条序列,总计约 1,440 亿个核苷酸。所有序列均从完整基因组中提取,并划分为最长 8,192 个核苷酸的非重叠片段。
在评估方面,研究人员从三个互补维度构建测试集,以全面考察模型能力。首先,在局部编码适合度层面,采用 MaveDB 中来源于大肠杆菌 K12 的 12 组核苷酸级实验数据,共计 21,250 个数据点,用于评估模型对局部编码语法及蛋白质适合度景观的刻画能力。
其次,在全基因组尺度的功能判断上,依托必需基因数据库(DEG),为 62 种细菌构建二元必需性标签。相关序列与注释均来自 NCBI,并以与 DEG 条目名称及序列一致性大于 99% 作为必需基因标注标准,最终形成 185,226 个数据点,用于评估模型整合长程依赖与基因组上下文的能力。
最后,在结构可解释性方面,以枯草芽孢杆菌基因组为例,结合其功能注释,将序列划分为不同功能区域,通过分析模型分段结果与真实生物学结构的对齐程度,验证其结构建模能力。
dnaHNet 模型:无需分词器的自回归前沿模型
dnaHNet 是一种无需显式分词器的基因组基础模型,其关键在于通过「动态分块」机制,让模型自行学习序列中的结构单元。这一设计既避免了固定分词对生物学功能片段的割裂,也缓解了逐核苷酸建模的计算开销,从而在表达能力与计算效率之间取得更优平衡。
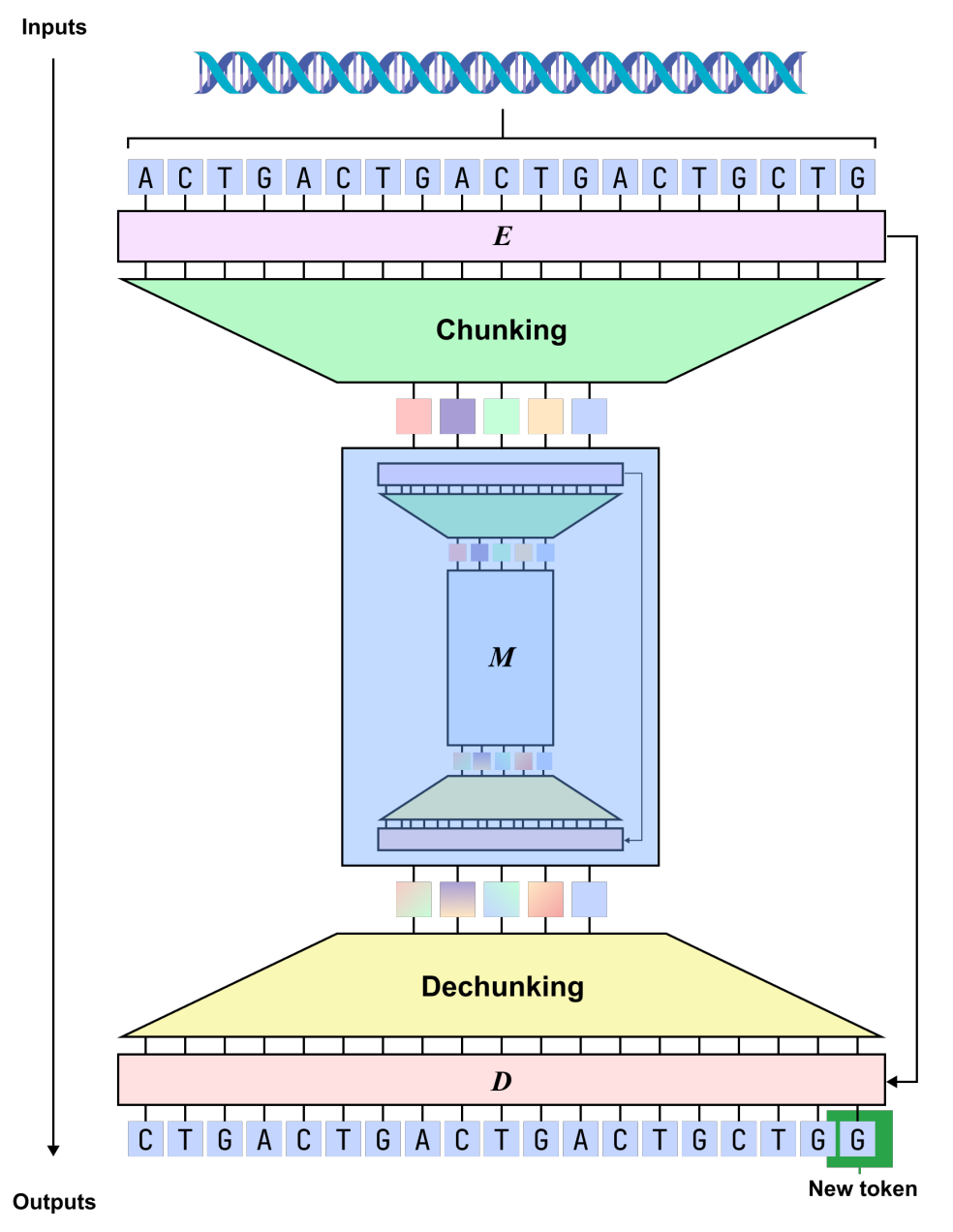
在建模形式上,dnaHNet 将基因组学习统一为自回归序列预测任务,即基于已有上下文预测下一个核苷酸。整体采用分层架构,每一层由编码器、主干网络和解码器构成:编码器通过路由机制识别序列中信息变化显著的位置(如密码子边界或调控区域),并据此将序列压缩为隐式分块表示;主干网络采用 Mamba 与 Transformer 结合的混合结构,对压缩序列进行建模,以兼顾长程依赖与关键信息交互;解码器再通过上采样将表示还原至核苷酸分辨率,输出预测结果。
在此基础上,dnaHNet 针对基因组数据进行了多项关键优化。首先,在参数分配上,约 30% 的模型容量用于编码器与解码器,以增强对局部结构的刻画能力。
其次,在结构设计上引入双阶段分层压缩:第一阶段侧重捕捉短尺度模式(如密码子),第二阶段建模更长范围的功能结构,从而在压缩效率与信息保真之间取得平衡。此外,在训练过程中结合自回归预测损失与压缩率约束,使模型在保证预测精度的同时,实现对计算开销的有效控制。
在推理阶段,模型依据边界概率动态决定分块方式,使建模粒度能够随上下文自适应变化,从而更贴近真实的基因组结构。
dnaHNet 计算成本降 3.89 倍,多任务表现领先
为系统评估 dnaHNet 的性能,该研究将其与 StripedHyena2 和 Transformer++ 两类主流长序列基因组模型进行对比,实验涵盖缩放特性、零样本变异效应预测、基因必需性预测以及生物学结构建模等多个方面。
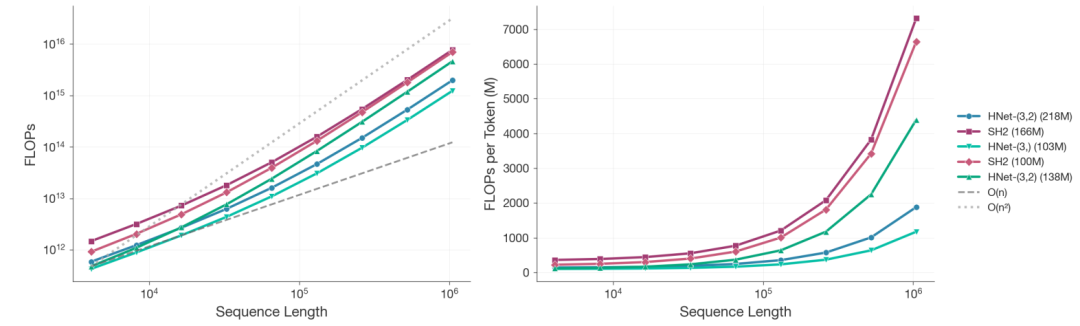
在缩放分析中,研究人员在固定算力预算下训练了超过 100 个不同规模的模型。当序列长度达到 10⁶ 核苷酸、总算力为 8×10¹⁹ FLOP 时,218M 参数的 dnaHNet 相比 166M 参数的 StripedHyena2 计算成本降低约 3.89 倍,双阶段结构的效率进一步优于单阶段版本。
基于困惑度的幂律拟合结果显示,在达到相同性能水平时,StripedHyena2 所需算力约为 dnaHNet 的 3.75 倍。此外,dnaHNet 的最优数据—参数配置明显偏离传统缩放定律:在相同算力条件下,其训练令牌数可达 140B,而对比模型仅为 68B 且尚未收敛。
在下游任务中,dnaHNet 在零样本蛋白质变异效应预测(MaveDB)与基因必需性预测(DEG)上均持续优于对比模型,且优势随算力提升而进一步扩大。这表明其动态分块机制与分层架构,能够更有效地整合局部编码语法与全局上下文信息,从而提升对生物功能的刻画能力。
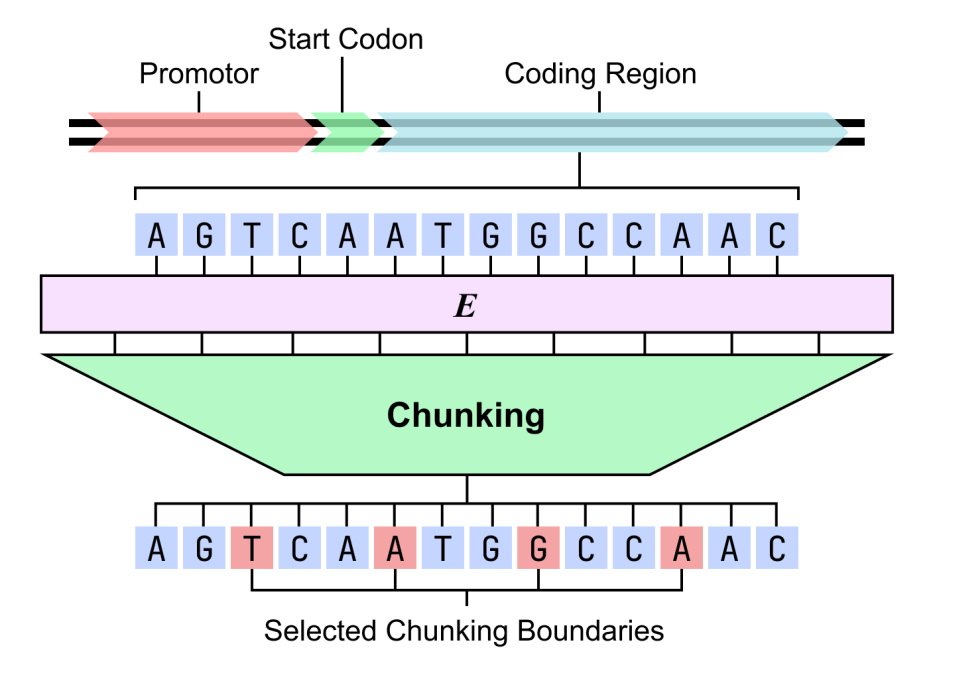
在结构可解释性方面,以双阶段 dnaHNet 对枯草芽孢杆菌基因组进行分析,结果显示模型能够自发学习具有生物学意义的分层结构:第一阶段呈现出对密码子的敏感性,能够准确捕捉编码区的三联体模式;第二阶段则更关注功能层面的结构,启动子、起始密码子及基因间区的分段概率显著高于编码区。
这一结果表明,模型不仅具备高性能预测能力,还能够在无监督条件下重建基因组的功能组织结构,从而为「DNA 语法」的解析提供了可解释的计算路径。
结语
整体而言,dnaHNet 不再预设序列的切分方式,而是让模型自己学习。实验证明,这种动态分块的分层建模既提升了计算效率,也更贴合基因组的多尺度结构。长远来看,如果能稳定学到有意义的生物学单元,就有望揭示基因组中难以形式化的规律,为变异预测、功能发现和合成设计等研究打开新空间。