Command Palette
Search for a command to run...
基于 1.4 万真实数据,华盛顿大学/微软等提出 GigaTIME,绘制全景肿瘤免疫微环境图谱
在癌症的演进图景中,肿瘤免疫微环境不仅主导着癌细胞的生长、侵袭与转移,也深刻影响着治疗反应与患者的最终预后。这并非一场癌细胞的「独角戏」,而是一个高度动态的生态系统——免疫细胞、成纤维细胞、内皮细胞等各类角色在此交织互动,共同嵌入结构与功能均已重塑的细胞外基质,形成一张精密而复杂的病理网络。
解析这张网络的关键,在于读懂细胞的功能状态与相互作用,而特定蛋白的激活水平正是其中重要的「分子密码」。传统上,免疫组织化学(IHC)技术以其直观显示蛋白定位的能力,成为破译密码的经典工具。例如,PD-L1 染色已被广泛用于识别免疫检查点状态,以预测免疫治疗疗效。然而,IHC 一次仅能捕获一种蛋白信息,难以还原多蛋白共存的真实生态,这构成了深入理解肿瘤—免疫细胞对话机制的主要瓶颈。
为突破这一局限,多重免疫荧光(mIF)技术应运而生。它能在一张组织切片上同步呈现多种蛋白的空间分布,完整保留组织结构的上下文信息。然而,该技术成本高昂、流程繁琐,从染色、成像到分析都极为耗时,导致大规模数据积累艰难,临床转化步履维艰。
与此形成鲜明对比的是,临床上广泛可得、成本低廉的 H&E 染色切片。它虽不能直接显示蛋白活性,却完整保留了组织的整体结构与细胞形态细节。其中隐藏的特征,或许正间接映射着细胞的功能状态,只是这些细微而复杂的模式,往往超出了人眼的辨识极限。
近年来,人工智能技术的突破带来了新的转机。通过对海量病理图像进行预训练,AI 已展现出强大的视觉解析与特征挖掘能力。这引出了一个关键设想:能否借助 AI,从易获取的 H&E 图像中「解码」出原本需要昂贵 mIF 技术才能捕获的蛋白活化信息?
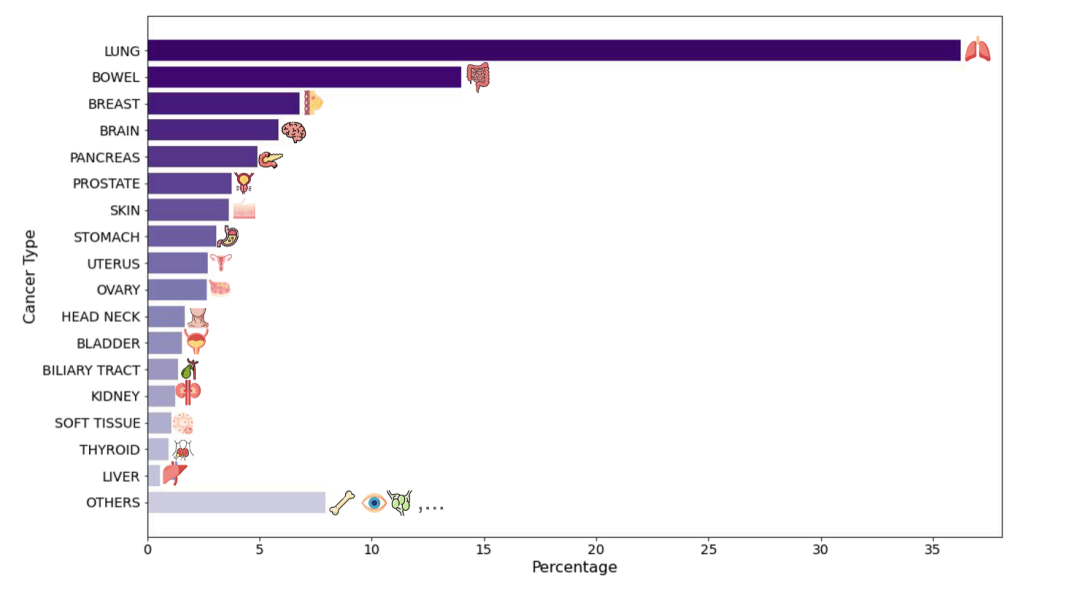
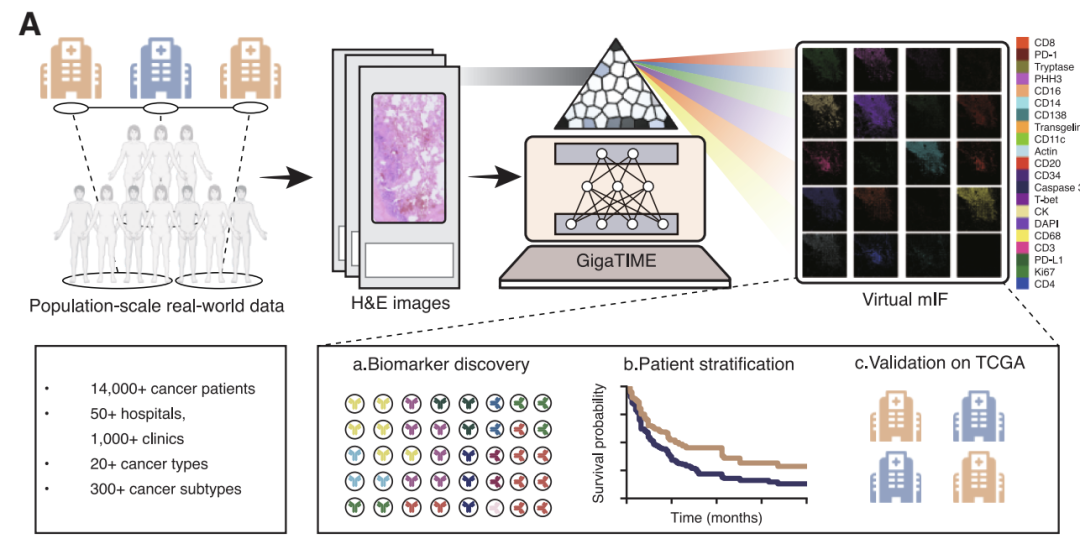
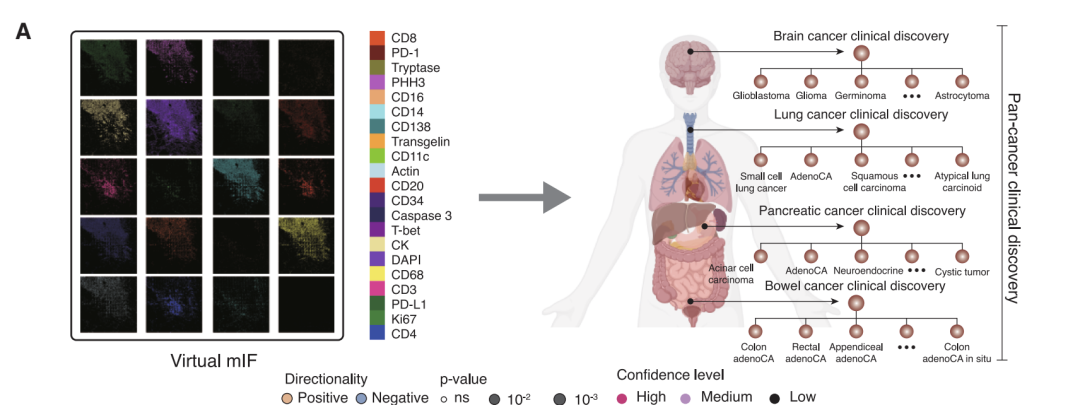
基于这一思路,由微软研究院、华盛顿大学与 Providence Genomics 组成的研究团队,提出了多模态人工智能框架 GigaTIME,其依托先进的多模态学习技术,能够从常规 H&E 切片生成虚拟的 mIF 图谱。研究团队将其应用于美国普罗维登斯医疗集团超过 14,000 名癌症患者的队列,涵盖 24 种癌症类型、 306 个亚型,最终生成了近 30 万张虚拟 mIF 图像,实现了对大规模多样化人群的肿瘤免疫微环境系统性建模。
相关研究成果以「Multimodal AI generates virtual population for tumor microenvironment modeling」为题,已发表于 Cell 。
研究亮点:
* GigaTIME 使用多模态 AI 将 H&E 病理切片转化为空间蛋白质组学数据,从常规 H&E 切片中生成包含细胞状态的虚拟人群。
* 支持大规模的临床发现和患者分层,并揭示了新的空间和组合蛋白质激活模式。

论文地址:https://www.cell.com/cell/fulltext/S0092-8674(25)01312-1
关注公众号,后台回复「多重免疫」获取完整 PDF
更多 AI 前沿论文:
https://hyper.ai/papers
数据集:构建从训练到应用的完整闭环
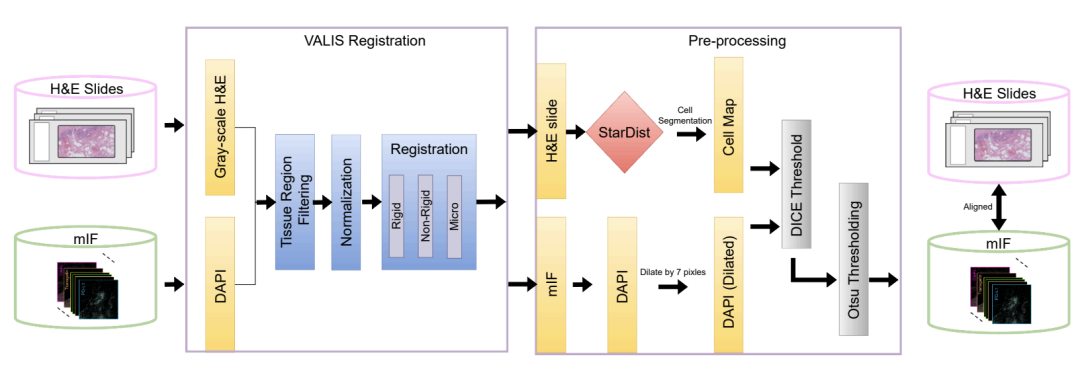
训练模型首先要解决一个根本矛盾:临床上普及且低成本的 H&E 染色无法直接显示蛋白质活性,而能揭示多蛋白空间关系的 mIF 技术,却又因昂贵复杂而难以大规模开展。为了构建连接这两种成像技术的 AI 模型,研究团队借助 COMET 平台,从 21 张 H&E 染色切片中采集了 441 张 mIF 图像。如下图所示,这些图像覆盖了从 DAPI 、 PHH3 等核蛋白,CD4 、 CD11c 等表面蛋白,到 CD68 等胞质蛋白,共计 21 个关键生物标志物,为解析肿瘤微环境中免疫细胞组成、功能状态及肿瘤细胞活动提供了重要依据。

获得配对图像后,更大的挑战在于如何从中提取高质量的训练数据。为此,如下图所示,团队设计了一套严谨的处理流程:首先使用 VALIS 工具,在像素级别上将 H&E 图像与 mIF 图像精确对齐;接着运用 StarDist 算法,对图像中的每个细胞进行识别与切割;最后依据 Dice 系数筛选出配准质量最高的图像区域。
经过层层质量控制,团队从包含 4 千万个细胞的初始数据中,精选出 1 千万个高质量细胞,并划分为训练集、验证集和独立测试集。此外,研究还引入了来自组织微阵列的乳腺癌与脑癌样本作为外部验证集。这些样本在组织结构和形态上与训练数据存在明显差异——它们呈现为由空白区域分隔的圆柱形小组织块,而非训练数据中大片连续的组织切片,从而有效检验了模型面对新样本类型和未见癌种时的泛化能力。
在模型应用层面,研究构建了两个规模宏大且相互补充的虚拟人群队列。第一个队列来自美国 Providence Health 医疗集团的临床网络,包含旗下 51 家医院和 1,000 多家诊所的 14,256 名癌症患者的 H&E 切片,覆盖 24 种主要癌症类型和 306 个细分亚型,同时整合了基因组标志物、病理分期和生存随访等丰富的临床信息。这一数据集的独特价值在于其真实世界特征:患者群体高度多样化,疾病阶段横跨早期到晚期的完整谱系,能够真实反映临床实践中的复杂情况。
第二个队列则取自公共数据库的癌症基因组图谱(TCGA),包含 10,200 例以早期、未经治疗的手术样本为主的 H&E 切片。这两个队列在患者来源、疾病阶段和临床背景上形成了鲜明对照。这种差异化设计,为验证模型的可靠性和普适性提供了绝佳条件:如果模型能在如此不同的数据集中得出一致且稳健的生物学结论,将有力证明其具备广泛的临床应用潜力。
GigaTIME 搭建形态与功能的智能桥梁
GigaTIME 模型直面肿瘤免疫微环境研究的关键瓶颈:高成本、低通量的 mIF 技术难以普及,而临床常规的 H&E 染色图像又无法直接反映蛋白质功能活性。该模型通过人工智能,学习从 H&E 图像生成虚拟的 mIF 图像,从而为在人群尺度上低成本、系统性地解析肿瘤免疫微环境提供了可行路径。
模型采用了一个经过精心设计的补丁式编码器-解码器框架,其核心是基于嵌套 U 型网络构建的。这种架构的优势在于能够同时捕获图像的局部细微特征和全局组织结构。具体来说,网络的编码器部分通过卷积与下采样操作,逐步从输入的 256×256 像素 H&E 图像块中提取多层次的特征表示;解码器部分则通过上采样与特征融合,将这些抽象特征重构为具有空间分辨率的虚拟 mIF 图像。这种设计使得模型既能关注单个细胞的精细形态,又能理解细胞群体的组织模式。
在输出层面,模型的设计体现了对生物学问题的深入考量。针对 21 个预设的蛋白质通道,GigaTIME 会对输入图像中的每一个像素进行二元分类预测,判断该位置是否存在特定蛋白的激活,最终生成像素级别的蛋白质活性图谱。这些局部的预测结果可以无缝拼接,还原出整张组织切片的虚拟 mIF 图像,进而支持计算各种定量指标,如特定蛋白在肿瘤区域的激活密度、空间分布模式等,为后续的高通量分析与临床关联研究提供坚实的数据基础。
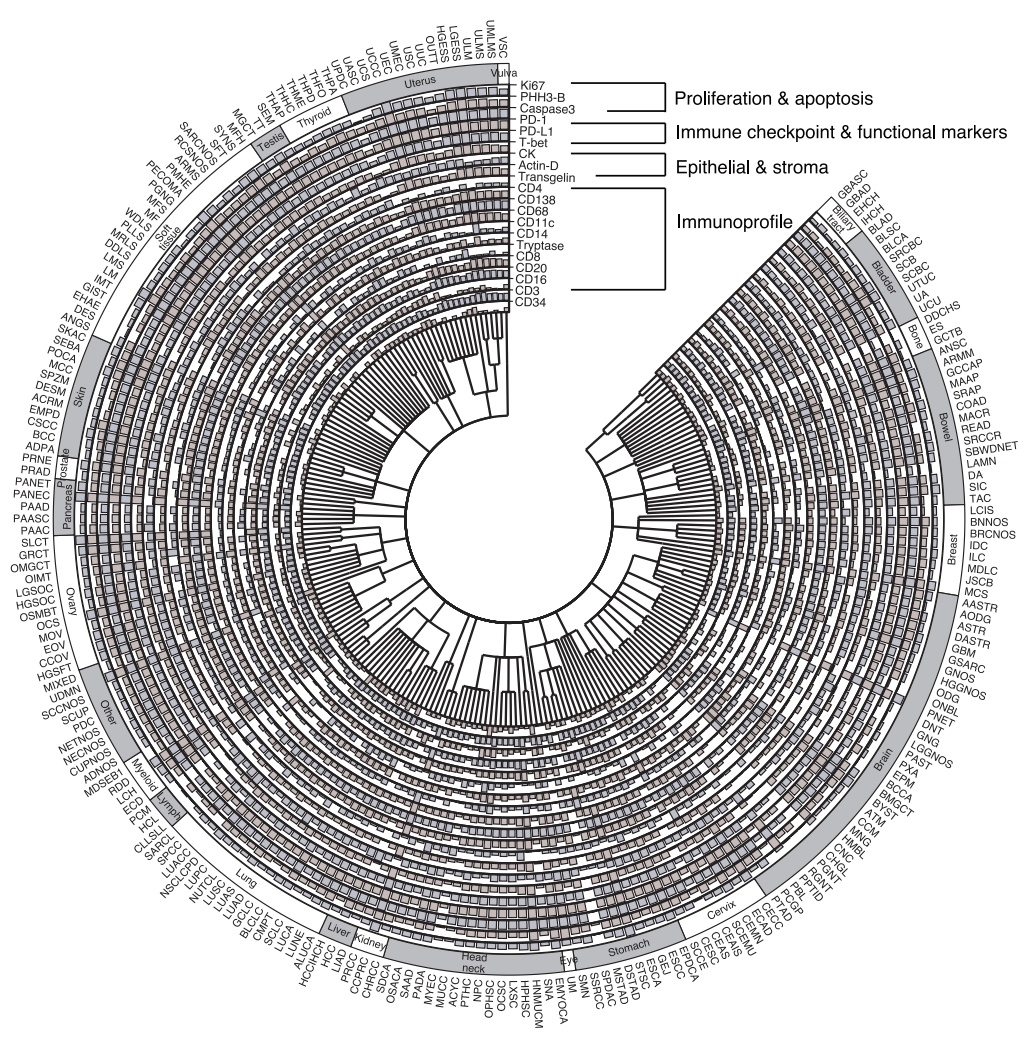
为确保模型有效学习,训练策略经过了系统优化。损失函数巧妙结合了 Dice 损失与二元交叉熵损失:前者侧重于保证预测活性区域与真实区域在空间轮廓上的整体一致性,后者则专注于提升每个像素点分类的精确性,两者的协同作用既保证了全局空间模式的准确还原,又确保了细节层面的可靠性。模型在 8 块 NVIDIA A100 GPU 上进行了 250 个 epoch 的充分训练,批处理大小为 16,学习率为 0.0001,所有关键超参数均基于验证集效果经过系统调试确定。
需要特别强调的是,模型的成功极大地依赖于高质量的训练数据。研究团队通过严格的图像配准、细胞分割和质量控制流程,从海量初始数据中精选出 1,000 万个高质量细胞用于训练,确保了模型所学到的是稳健、可靠且具有生物学意义的跨模态映射关系,而非表面的统计规律或噪声模式。
基于近 30 万虚拟图像的规模化发现:GigaTIME 揭示 1234 个临床关联
为全面评估 GigaTIME 的性能与价值,研究团队从技术验证与临床发现两个维度设计了系统的评估方案。
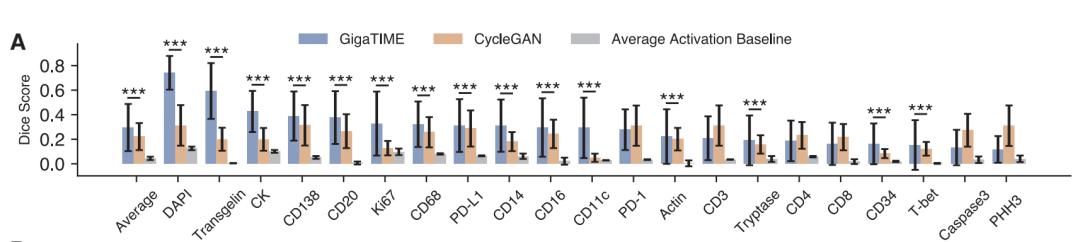
在技术验证上,研究从像素、细胞和切片 3 个层面评估模型的图像转换能力。在像素层面,GigaTIME 在 21 个蛋白通道中的 15 个上显著优于基线模型 CycleGAN 。例如在 DAPI 通道上,GigaTIME 的 Dice 系数达到 0.72,远超简单统计基线的 0.12 。
在细胞层面,GigaTIME 在 DAPI 通道的相关性达到 0.59,而 CycleGAN 仅为 0.03,接近随机水平。
在切片层面,GigaTIME 的 DAPI 通道相关系数高达 0.98,所有通道平均为 0.56,而 CycleGAN 均接近 0 。这些结果证明,基于高质量配对数据的有监督训练对于准确的跨模态转换至关重要。
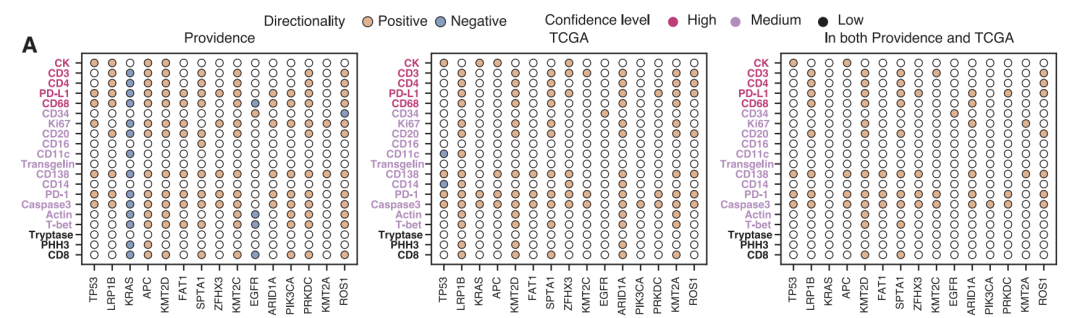
在临床发现方面,研究利用 14,256 名患者的近 30 万张虚拟 mIF 图像,系统分析了虚拟蛋白表达与 20 种临床生物标志物的关联。经严格的统计检验与多重校正,共识别出 1,234 个显著关联,分布在泛癌、癌种、癌亚型三个层面。
在泛癌分析的 175 个关联中,高肿瘤突变负荷和高微卫星不稳定性与多种免疫浸润标志物(CD138 、 CD20 、 CD68 、 CD4)的激活增强显著相关,符合抗原驱动免疫激活机制。同时发现了新线索:KMT2D 突变与免疫标志物呈强正相关,提示可能促进免疫浸润;而 KRAS 突变与免疫标志物呈负相关,体现免疫排斥表型。在特定癌种及亚型中,模型揭示了大量特异性关联。例如脑癌中 T-bet 与 TP53 突变的强相关在泛癌层面未被检测到,可能与中枢神经系统独特的免疫微环境相关。肺癌亚型分析显示,肺腺癌中 PRKDC 突变与免疫响应标志物的关联强于肺鳞状细胞癌,印证了结合组织学背景解读的重要性。
研究还验证了虚拟数据在临床预后中的价值。分析发现,原发肿瘤大小(T 分期)与免疫检查点及浸润标志物呈正相关,但这一关联在疾病进展到晚期时发生反转,提示晚期肿瘤可能主要通过其他免疫逃逸机制驱动。在生存分析中,整合全部 21 个通道的复合特征在患者分层上优于单一蛋白,充分体现了多参数联合分析的价值。
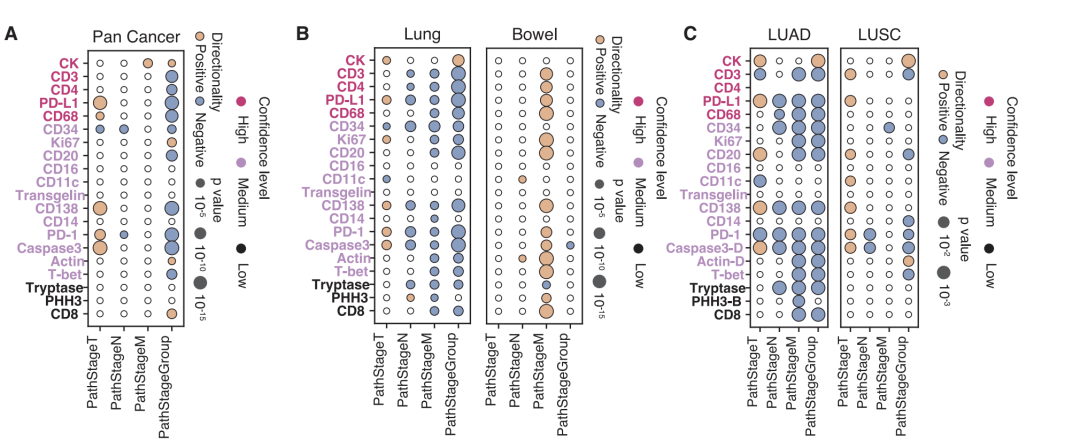
为确保可靠性,所有主要发现在 TCGA 的独立队列中进行了验证。尽管两个人群在来源和临床特征上差异显著,核心发现仍高度一致(癌亚型层面斯皮尔曼相关系数达 0.88),共同发现的 80 个显著关联表现出极高的统计学富集(p<2×10⁻⁹)。同时,Providence Health 虚拟人群在泛癌层面发现的显著关联比 TCGA 多 33%,凸显了大规模真实世界数据的独特价值。
探索性分析还揭示了复杂空间模式的价值。熵、信噪比、锐度等指标分别在 89 个、 63 个、 79 个蛋白-生物标志物配对中表现优于简单的激活密度。研究还发现了蛋白间的协同作用:CD138 与 CD68 的组合在预测 20 种生物标志物中优于单一蛋白,其中 13 种具有统计学差异,提示浆细胞与巨噬细胞可能通过抗体介导机制协同对抗肿瘤。
AI 赋能:从虚拟蛋白图谱到肿瘤研究新前沿
通过 AI 从常规病理切片生成虚拟空间蛋白质组学图像,正位于数字病理与计算生物学创新的核心。这一方向吸引了全球顶尖学术机构的探索,也催生了生物科技企业的商业实践。
在学术界,斯坦福大学在 Nature Medicine 发布的 HEX 模型,通过 819,000 个配对图像区块训练,能预测 40 种生物标志物的空间表达,展现出比 GigaTIME 更广的蛋白覆盖。加州大学旧金山分校在 Science Translational Medicine 发布的 DeepHeme 系统,以近 5 万张多中心高质量数据为基础,实现了 23 类骨髓细胞的精准分类,为血液病诊断的自动化提供了范例。
在产业界,Reveal Biosciences 获比尔及梅琳达·盖茨基金会支持,开发从病理图像提取「数字生物标志物」的平台,加速全球健康研究。另一路径是通过硬件创新降低成本,如 Micronit 开发的微流控设备大幅降低样本和试剂消耗。 Optellum 的肺结节诊断平台获 FDA 批准,为从常规数据中挖掘深层特征用于临床决策提供了商业化范式和监管先例。
GigaTIME 作为这一领域的重要里程碑,不仅展示了多模态 AI 在肿瘤免疫微环境研究中的巨大潜力,更为后续研究提供了可复用的技术框架和数据资源。未来发展将依赖于「虚拟-现实」融合的数据生成能力与低成本检测技术的共同进步,最终为理解肿瘤复杂性、加速精准医疗带来变革性工具。
参考链接:
1.https://mp.weixin.qq.com/s/AsqSemP3idCbIJ7xQ3gXGg
2.https://mp.weixin.qq.com/s/umg-UrMm6Qe-R-MbLpLZOQ








