Command Palette
Search for a command to run...
耶鲁大学提出 MOSAIC,构建超 2 千个 AI 化学专家,专业分工高效锁定最优合成路线
现代合成化学正面临知识迅速积累与应用转化效率之间的突出矛盾。每年有数十万篇相关文献发表,可用合成知识的总量已积累至百万级别。然而,这些知识大多以非结构化文本形式分散在不同数据库中,呈现出显著的碎片化特征。依赖传统的文献检索与人工筛选,不仅过程耗时费力,也难以系统覆盖跨领域的反应类型,导致大量潜藏在文献中的有效信息难以被提取并转化为可执行的实验方案。
面对这一知识管理困境,合成实践的核心需求日益聚焦于如何高效获取高可重复性的完整实验流程。这类流程涉及试剂选择、化学计量控制、温度程序和后处理步骤等诸多关键参数。
目前,该领域的发展主要受限于两方面,其一是专家经验难以覆盖持续扩张的反应空间,在跨学科合成任务中往往伴随高昂的试错成本;其二,尽管人工智能技术发展迅速,但通用模型在化学领域的应用仍存在可靠性不足、易产生「幻觉」及缺乏置信度评估等问题,尚无法满足实验级的精度要求。因此,将海量、分散的化学知识转化为结构化、可信赖的合成指导,已成为突破领域效率瓶颈的关键。
在此背景下,耶鲁大学研究团队近期提出 MOSAIC 模型,将通用大语言模型转化为一个由众多专业化学专家构成的协作系统,通过专业分工有效抑制模型幻觉,提供可量化的不确定性评估,实现了从反应描述到完整实验方案的系统生成,有望在药物发现、材料开发等领域实质性地提升科研效率。
相关研究成果以「Collective intelligence for AI-assisted chemical synthesiss」为题,已发表于 Nature 。

论文地址:
https://www.nature.com/articles/s41586-026-10131-4
关注公众号,后台回复「MOSAIC」获取完整 PDF
更多 AI 前沿论文:
基于 Pistachio 数据库,构建各擅所长的「AI 化学专家」
该研究基于 Pistachio 数据库开展。这是一个商业化、高度结构化的化学反应知识库,内容主要源自全球专利文献。通过对专利中记载的反应物、产物、试剂、溶剂、产率及关键步骤文本描述进行系统提取与标准化处理,数据库将其统一编码为机器可读的格式(如 SMILES 字符串)。研究团队并未直接使用全量数据,而是执行了严格的质量筛选,核心标准是要求反应记录必须包含详细、可执行的实验步骤描述,而不仅仅是反应物与产物的映射关系,从而确保后续训练的模型学习的是「如何实现反应」,而非仅仅是「反应结果是什么」。
经过筛选的数据通过专门设计的核度量网络,被转化为 128 维的反应特异性指纹。该数字化表征旨在捕捉化学反应的本质转化特征,所有指纹向量共同构成了一个表征广阔化学知识空间的「反应宇宙」。基于此向量空间,研究采用无监督的 Voronoi 聚类算法(通过 FAISS 库实现),将其划分为 2,489 个互不重叠的专业区域,每个区域聚集了化学性质高度相似的反应类型。
最终,每个 Voronoi 区域内的反应文本被用于独立微调一个专用的 Llama-3.1-8B-Instruct 模型,由此形成 2,489 个各擅所长的「AI 化学专家」。整个 MOSAIC 框架的知识范围与能力边界,根本上由这份以专利为核心的训练数据集所决定。这也解释了系统在某些快速发展的前沿领域(如光化学)表现相对受限的原因——这些内容在现有专利数据库中的覆盖尚不充分。
MOSAIC:由众多专业化学专家构成的去中心化协作系统
MOSAIC 模型的核心设计思想是将通用大语言模型 Llama-3.1-8B-instruct 转化为一个由众多专业化学专家构成的去中心化协作系统。这一搜索驱动的架构显著降低了对硬件资源的需求,仅需使用适中规模的算力配置(如 4 个 GPU)即可对特定任务子集进行训练,无需依赖大规模计算集群。系统通过专家分工机制有效抑制模型幻觉,并提供可量化的不确定性评估,同时支持动态扩展新专家而无需重新训练整个系统,在灵活性与可持续性上具有明显优势。
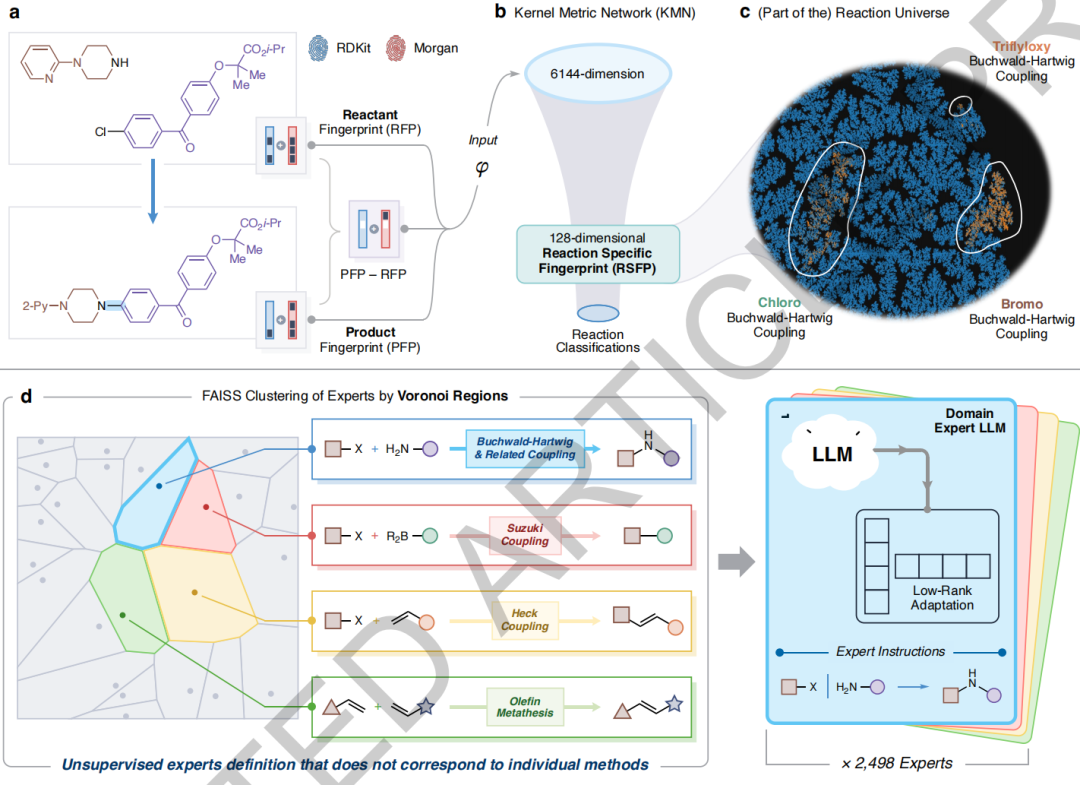
为突破大语言模型在大规模数据上训练时面临的算力与协调瓶颈,MOSAIC 通过 3 个渐进式组件构建而成:
反应相似性度量:
研究设计了一种基于神经网络的非线性映射(核度量网络,KMN),用于量化化学反应之间的相似性。它将 SMILES 编码的反应转化为 128 维的反应特异性指纹(RSFP),使其欧氏距离能够近似反应类别关系,从而捕捉反应的本质转化特征。
知识空间聚类:
利用 FAISS 库的高效索引能力,对 RSFP 向量空间进行无监督 Voronoi 聚类,自动划分出 2,498 个化学性质高度聚集的专业区域,每个区域代表一个特定的化学知识领域。
领域专家训练:
在每个聚类对应的反应数据上,独立微调专用的专家模型。研究采用两阶段训练策略:先在完整数据集上进行基础模型微调,再利用各聚类数据深化对应专家的领域知识,使专家在保持通用化学理解的同时,具备深厚的专业认知。
MOSAIC 首先将查询反应编码为 RSFP,并通过 FAISS 快速定位其所属的 Voronoi 区域及对应的专家。例如,对于一个氯代芳烃的 Buchwald-Hartwig 偶联反应,系统会调用精于此领域的专家,生成完整、可读的合成步骤。实验验证表明,完全依照该方案操作,最终能以 96% 的产率获得目标产物。
MOSAIC 实现 94.8% 组分覆盖率与 71% 合成成功率
该研究进一步通过多维度评估系统验证了 MOSAIC 模型的综合性能,其核心价值在于将海量文献知识转化为高可信度的合成智能。
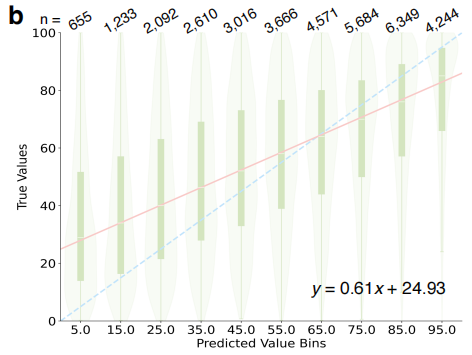
在产率预测与核心组分识别方面,MOSAIC 模型通过解析完整的实验程序文本,实现了对反应产率的量化预测。如下图所示,采用分箱策略后,预测区间中心与真实产率中位数显示出显著相关性(R² = 0.811)。在识别反应关键组分(试剂、溶剂)方面,模型展现出优秀的覆盖能力,在集成前三位专家的预测结果后,能至少部分识别出正确组分的综合成功率高达 94.8% 。值得注意的是,即便预测条件与文献记录不完全一致,其输出也常为化学上可行的替代方案,体现了深层的专业判断力。
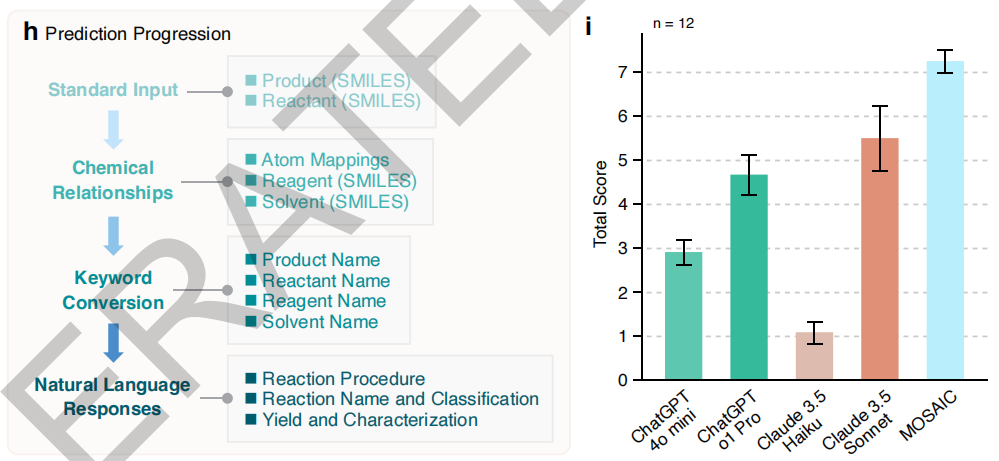
在 12 类重要反应(Suzuki 偶联、 Buchwald-Hartwig 胺化等)的对比测试中,如下图所示,与 ChatGPT-4o 、 Claude 3.5 等通用大语言模型相比,MOSAIC 在提供明确、可行的合成指导方面 consistently 表现更优。这一优势在模型参数量仅为 80 亿的背景下尤为突出,证明了领域专业化微调的有效性。更重要的是,MOSAIC 克服了通用模型在化学任务中常见的指令遵循不稳定、回答随意性大等问题,提供了稳定、可靠的输出,这对实际实验至关重要。
为了评估所提出框架的实用性、通用性和可靠性,该研究还通过执行现代化学合成基础反应的精确、最高排名预测进行了广泛的实验验证。研究人员把重点放在了对药物和材料开发至关重要的广泛适用的催化反应上。 Buchwald-Hartwig 胺化形成的碳-氮键在药物分子中普遍存在,这些具有挑战性的反应的条件被准确预测。实现了类药支架的高效组装,在对从天然产物到功能材料的应用至关重要的烯烃转化方面展示了特别的优势。
此外,MOSAIC 模型的实用性在大量新颖化合物的成功合成中得到有力证实。在总计 37 个目标化合物的合成中,有 35 个依据模型的首次推荐即告成功,整体成功率达 71% 。验证范围涵盖从经典偶联反应到选择性转化,并包括指导开发全新氮杂吲哚成环方法这一体现创新能力的案例。
尤为重要的是,模型内部的置信度指标(最近专家质心距离)与实验成功率呈现明确的正相关关系:高置信度预测(距离<100)的成功率超过 75% 。这为化学家提供了宝贵的量化决策依据,使其能在高成功率目标与探索性尝试之间进行有效的资源分配。
化学合成迈入精准智造新纪元
在推动化学合成智能化的全球进程中,学术界与工业界正沿着互补的轨道协同发力,共同重塑从分子发现到工艺生产的全链条。
高校的研究如同探索未知领域的先锋,专注于攻克底层计算的极限与科研范式的革新。麻省理工学院(MIT)的研究人员巧妙地将用于图像生成的「扩散模型」迁移至化学反应领域,实现了对关键「过渡态」结构的超快速计算——将传统需耗时数日的任务压缩至数秒之内,并以 0.08 埃的原子级精度为反应预测提供了前所未有的微观洞察。
与此同时,斯坦福大学的团队则致力于重构科研本身的工作方式,构建 AI 驱动「虚拟实验室」系统,能够自主组建多学科虚拟团队,在「首席研究员 AI」的协调下进行秒级协作与辩论,已在疫苗设计等复杂课题中提出了超越常规的创新思路。此外,哈佛大学等机构的研究将人工智能的模拟能力推向宏观尺度,其提出的统一框架成功实现了对包含百万原子的复杂铁电材料的精确模拟,为从本质上设计新一代功能材料提供了强大的数字透镜。
相较于学术界对前沿的开拓,企业界的创新则更注重于将尖端算法转化为解决实际痛点的生产力与市场竞争力。德国化工巨头巴斯夫在全球范围内部署 AI,不仅推出了辅助研发的「AI 化学家 Copilot」,将新材料开发周期大幅缩短了 60%,更将 AI 深度融入生产优化、物流规划和预测性维护等环节,实现了从实验室到工厂的全价值链增效。
而在制药领域,总部位于瑞士的诺华(Novartis)等药企正以「端到端」的方式拥抱 AI,通过与 Isomorphic Labs 、 Schrödinger 等专业公司的深度合作,将人工智能的应用贯穿于从全新靶点发现、化合物生成与安全性预测,到优化临床试验设计的每一个关键环节,显著提升了药物研发的确定性与成功率。
纵观这些跨越学术与产业的突破,化学研究——这门曾高度依赖个人经验与重复试错的传统学科,正在被数据与算法深刻重塑,稳步迈向一个可预测、可规划、可自动执行的精准科学新时代。从攻克疾病的创新药物,到助力可持续发展的绿色材料,化学合成智能化这场深远变革,正在为我们应对这个时代最紧迫的挑战,锻造出前所未有的核心能力。
参考文章:
1.http://edu.people.com.cn/n1/2025/0730/c1006-40532541.html
2.https://cen.acs.org/pharmaceuticals/drug-development/Q-Novartiss-biomedical-research-head/103/web/2025/01








