Command Palette
Search for a command to run...
L’équipe Du MIT a Proposé Le Modèle FASTSOLV, 50 Fois Plus Rapide Que Le Modèle Original, Pour Prédire La Solubilité Des Petites Molécules À N’importe Quelle température.

En chimie et en science des matériaux, la solubilité des solides organiques dans différents solvants est une propriété moléculaire essentielle, impactant l'ensemble de la recherche et de la chaîne industrielle. Pour les procédés de synthèse, un contrôle précis de la solubilité permet non seulement de sélectionner le solvant optimal et d'optimiser les conditions de réaction, mais aussi d'améliorer significativement le rendement et la pureté du produit, réduisant ainsi les coûts de production. En sciences de l'environnement, c'est un paramètre clé pour analyser la migration et le devenir de polluants tels que les substances per- et polyfluoroalkylées (PFAS) dans le sol et l'eau, fournissant ainsi une base scientifique pour la prévention et le contrôle de la pollution. Enfin, dans des procédés tels que la cristallisation et la séparation membranaire, la solubilité est une variable essentielle qui détermine le comportement des phases et l'efficacité de la séparation.
Cependant, les méthodes de détermination expérimentales traditionnelles présentent de nombreuses limites : elles sont non seulement chronophages et consommatrices de matériaux, mais sont également facilement perturbées par des facteurs tels que la forme cristalline des solides organiques et les impuretés, ce qui entraîne une exactitude insuffisante des données. Selon les recherches, l'écart type interlaboratoire de la solubilité dans l'eau (logS) atteint souvent 0,5 à 0,7 unité logarithmique, et dans les cas extrêmes, la différence entre les résultats de mesure peut même être décuplée. Bien que des méthodes empiriques d'addition de groupes, des modèles de chimie quantique et des méthodes d'apprentissage automatique aient été appliqués à la prédiction,Cependant, il existe souvent des problèmes de polyvalence insuffisante ou de difficulté à équilibrer la précision et l’efficacité de calcul.
Pour résoudre ce problème, une équipe de recherche du Massachusetts Institute of Technology a combiné des outils d’informatique chimique avec la nouvelle base de données de solubilité organique BigSolDB.Amélioré sur la base de l'architecture des modèles FASTPROP et CHEMPROP,Le modèle peut saisir simultanément des molécules de soluté, des molécules de solvant et des paramètres de température, et effectuer directement une formation de régression sur logS.
Dans un scénario d'extrapolation stricte des solutés, comparé aux modèles SOTA existants tels que Vermeire,Le RMSE du modèle optimisé a été réduit de 2 à 3 fois et la vitesse d’inférence a été augmentée jusqu’à 50 fois.Actuellement, l'équipe a nommé le modèle dérivé FASTPROP FASTSOLV et l'a publié en open source, fournissant un outil efficace et pratique pour la recherche scientifique et les applications industrielles connexes.
Les résultats de recherche pertinents ont été publiés dans Nature Communication sous le titre « Prédiction de la solubilité organique basée sur les données à la limite de l'incertitude aléatoire ».

Adresse du document :
https://www.nature.com/articles/s41467-025-62717-7
Suivez le compte officiel et répondez « Solubilité organique » pour obtenir le PDF complet
Conception d'un système de construction et d'évaluation d'ensembles de données piloté par BigSolDB
La source de données principale de cette étude est BigSolDB, qui collecte systématiquement des données de solubilité des solides organiques dans une variété de solvants organiques et dans différentes conditions de température proches de la limite de précipitation, fournissant un support clé pour la formation de modèles de prédiction généraux.
Pour atteindre l’objectif de recherche « d’extrapoler de nouveaux solutés sans aucune connaissance préalable », l’équipe de recherche a conçu un système rigoureux de formation-évaluation :Le modèle a été formé sur BigSolDB et testé indépendamment sur deux ensembles de données publics : SolProp et Leeds.Pour éviter de sous-estimer la difficulté d’extrapolation, comme le montre la figure ci-dessous, cette étude a d’abord supprimé tous les solutés de SolProp qui chevauchaient BigSolDB, et a introduit l’ensemble de données de Leeds avec un espace chimique plus large en complément.
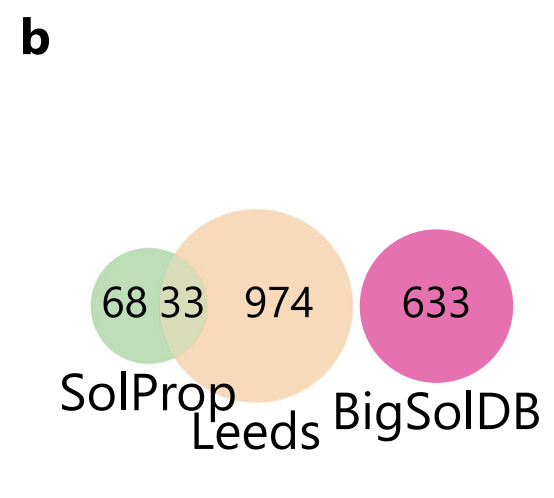
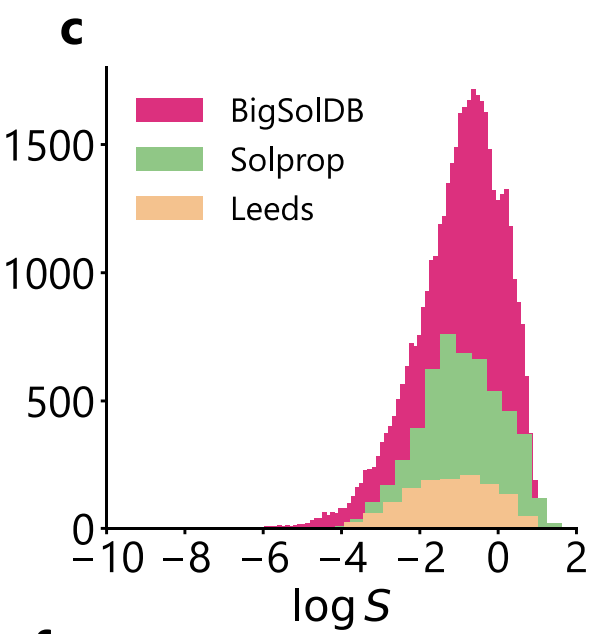
Comparé à SolProp,Leeds offre une plus grande diversité de solutés mais ne couvre que les conditions de température ambiante.Cela permet non seulement de tester l'adaptabilité du modèle à de nouveaux espaces chimiques, mais aussi d'augmenter la limite supérieure d'incertitude en raison de l'absence de réduction implicite du bruit due à la « moyenne multi-températures ». Notamment, comme le montre la figure ci-dessous, les distributions logS des trois ensembles de données sont très cohérentes, toutes concentrées à près de –1 et présentant une longue queue à l'extrémité de faible solubilité, ce qui garantit la comparabilité des distributions pour les comparaisons de performances entre les ensembles de données.
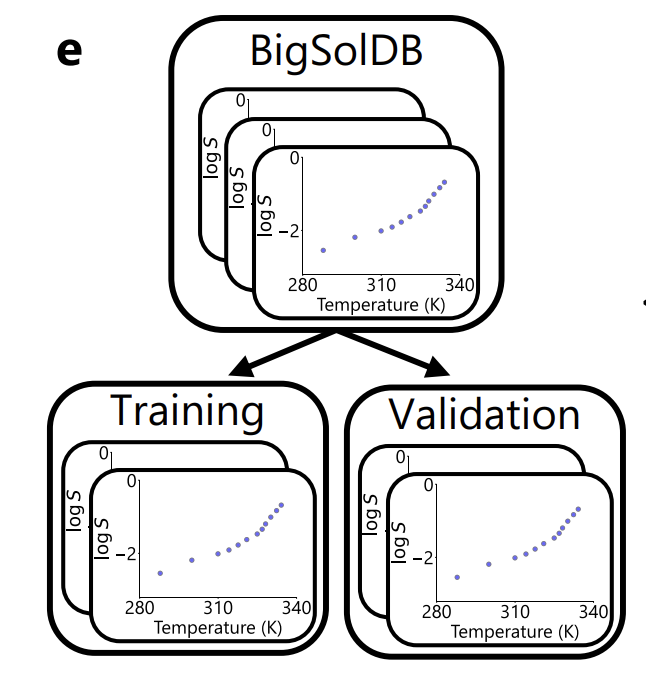
En termes de segmentation des données, comme le montre la figure ci-dessous, les chercheurs utilisent strictement le soluté comme unité : 95% de soluté sont utilisés pour l'entraînement, 5% sont utilisés pour la validation et la sélection du modèle,Toutes les mesures du même soluté dans différents solvants et à différentes températures n'apparaîtront pas dans différents sous-ensembles en même temps.Cela permet d’éviter efficacement les fuites d’informations.
De plus, l'étude a utilisé la boîte à outils ASTARTES pour diviser aléatoirement l'ensemble de validation en « expériences complètes » dans les données de formation, et a revérifié les limites de division des dimensions soluté et expérimentale dans l'évaluation finale pour garantir l'indépendance et la rigueur de l'évaluation.
Construction du modèle FASTSOLV pilotée par BigSolDB
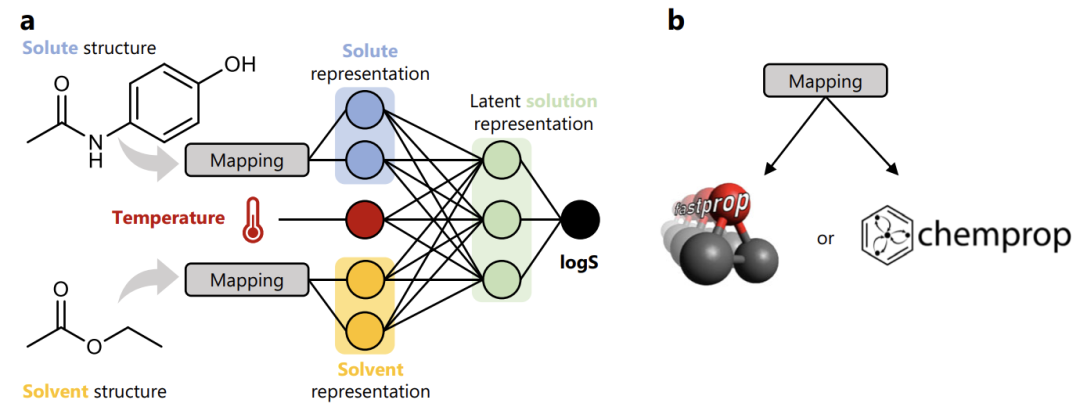
En s'appuyant sur l'ensemble de données BigSolDB, comme le montre la figure ci-dessous, cette étude a personnalisé les deux architectures de modèles classiques de FASTPROP et CHEMPROP et a construit un processus de modélisation d'apprentissage automatique clair.
d'abord,Cartographier les structures moléculaires des solutés (tels que le paracétamol) et des solvants (tels que l'acétate d'éthyle) dans les vecteurs de représentation correspondants ;Alors,Ces deux vecteurs de représentation moléculaire sont combinés avec le paramètre de température de la solution pour former une représentation de solution complète.final,La représentation a été introduite dans un réseau neuronal entièrement connecté et une formation de régression a été réalisée avec logS (logarithme de solubilité) comme cible.
Grâce à cette transformation, le modèle finalement développé parvient à une prédiction unifiée de la solubilité des petites molécules dans plusieurs solvants organiques + différents scénarios de température, brisant ainsi la dépendance du modèle traditionnel à des solvants ou des plages de température spécifiques.
Pour améliorer encore la robustesse et la fiabilité prédictive du modèle, l’équipe de recherche ne s’est pas appuyée sur une seule sortie de modèle.Au lieu de cela, le modèle FASTPROP est formé dans quatre conditions d'initialisation aléatoires différentes, puis le modèle FASTSOLV final est obtenu grâce à la combinaison de stratégies d'intégration.Toutes les analyses clés ultérieures, telles que les comparaisons de performances et les vérifications de cas, sont basées sur ce modèle intégré, réduisant ainsi efficacement le risque de fluctuation aléatoire d’un modèle unique.
Parallèlement, afin de mesurer objectivement les performances du nouveau modèle, l'étude a utilisé le modèle SOTA, désormais largement reconnu, le modèle Vermeire, comme référence de comparaison. Ce modèle est entraîné par quatre sous-modèles thermochimiques indépendants, puis produit des résultats de solubilité par combinaison de cycles thermodynamiques. Il présente l'avantage d'équilibrer la diversité des solvants et la dépendance à la température. Cependant, l'étude a révélé que l'ensemble de données SolProp utilisé pour ses tests présentait un chevauchement important de la structure des solutés avec son propre ensemble d'entraînement. Ce « chevauchement de données » peut entraîner une surestimation des performances extrapolées. Afin de garantir l'équité et la rigueur de la comparaison, cette étude a reproduit scrupuleusement la configuration d'entraînement-test originale du modèle Vermeire et a mené des expériences de contrôle sur cette base afin de garantir que la différence de performance est uniquement due au modèle lui-même et non aux conditions de test.
Mise à jour de l'extrapolation de la solubilité organique SOTA avec une précision 2 à 3 fois supérieure et une vitesse 50 fois supérieure
Cette étude a réalisé des tests multidimensionnels et vérifié les performances du modèle. Dans le scénario d'interpolation, le modèle FASTPROP optimisé a obtenu une RMSE = 0,22, P₁ = 94%, et le modèle CHEMPROP a obtenu une RMSE = 0,28, P₁ = 90%.Les performances ont approché le plafond de bruit des données expérimentales, confirmant la valeur de support de BigSolDB.
Dans le nouveau test d'extrapolation de soluté, comme le montre la figure ci-dessous, le modèle de Vermeire a obtenu de mauvais résultats sur l'ensemble de données de Leeds en raison d'une surestimation systématique (RMSE = 2,16, P₁ = 34%), tandis que les RMSE de FASTPROP et CHEMPROP ont chuté à 0,95 et 0,99 respectivement, et que P₁ a dépassé 69%. Sur l'ensemble de données SolProp, notre modèle a également obtenu de meilleurs résultats (RMSE = 0,83, P₁ = 80%).Et la vitesse d’inférence de FASTPROP est environ 50 fois supérieure à celle du modèle Vermeire.Prend en charge l'analyse d'interprétabilité SHAP.
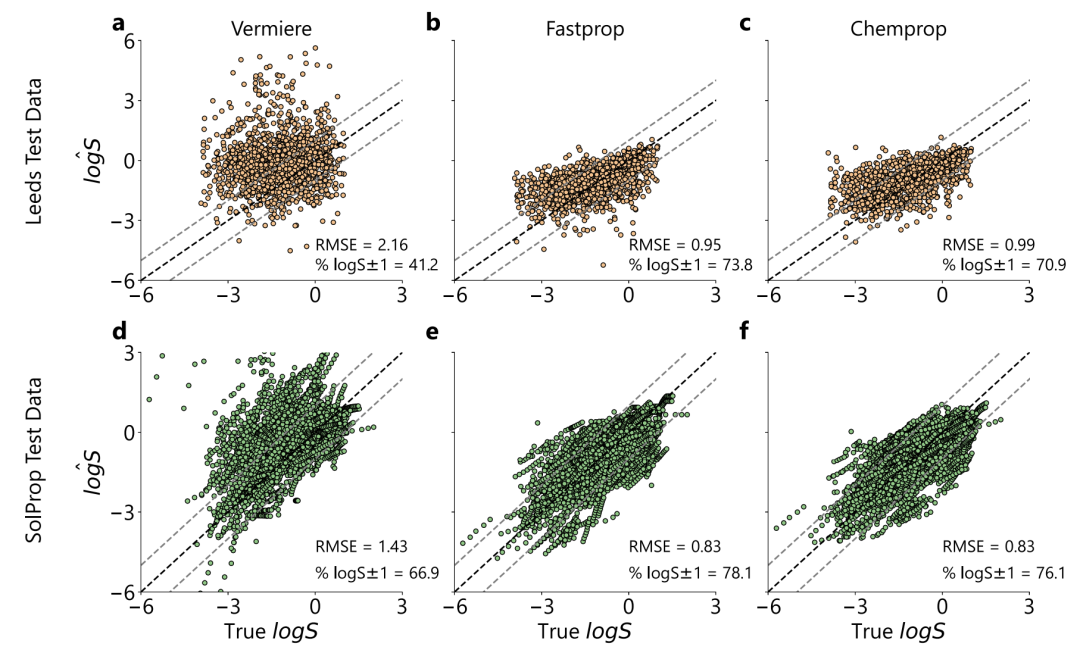
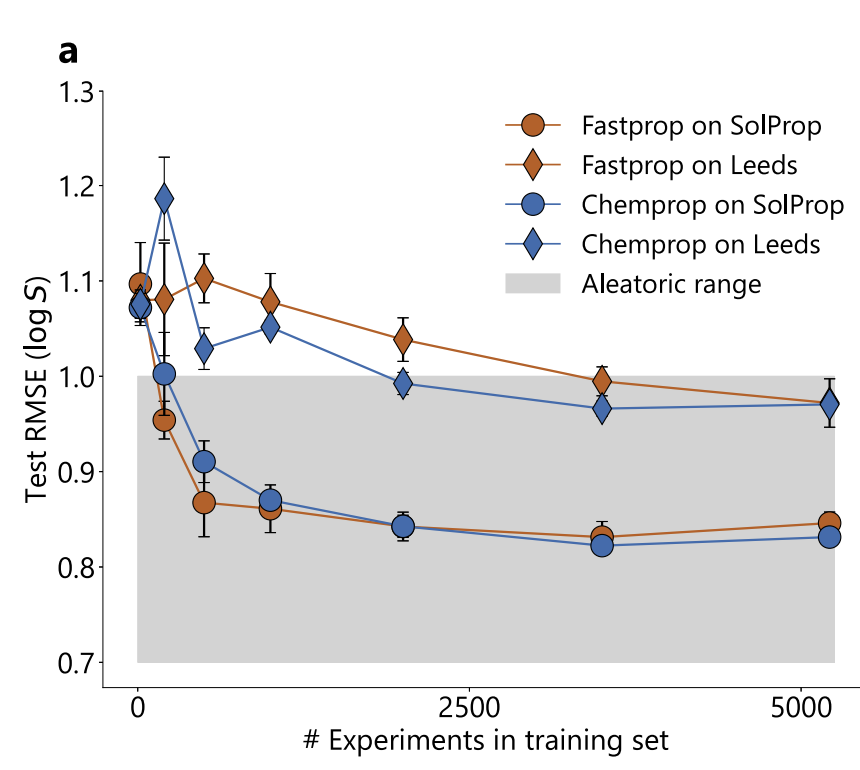
L'expérience de volume de données d'entraînement est illustrée dans la figure ci-dessous. Bien que FASTPROP et CHEMPROP aient des représentations moléculaires différentes, leurs performances convergent vers des limites similaires : l'ensemble de tests SolProp nécessite environ 500 expériences (environ 5 000 points de données) pour atteindre le plateau, tandis que CHEMPROP nécessite environ 2 000 expériences (environ 20 000 points de données) sur l'ensemble de tests Leeds.
Estimée à partir de 34 ensembles de données multi-sources dans les mêmes conditions dans BigSolDB, la limite d'incertitude aléatoire expérimentale est RMSE = 0,75 unités log, tandis que la RMSE des deux modèles sur SolProp est de 0,83, ce qui est proche de cette limite ; comparés à de grands modèles tels que MolFormer et ChemBERTa-2, les deux modèles fonctionnent mieux.Cela prouve que le goulot d’étranglement des performances provient des données expérimentales plutôt que de l’expressivité du modèle.
Test moyen des performances du modèle à des limites arbitraires
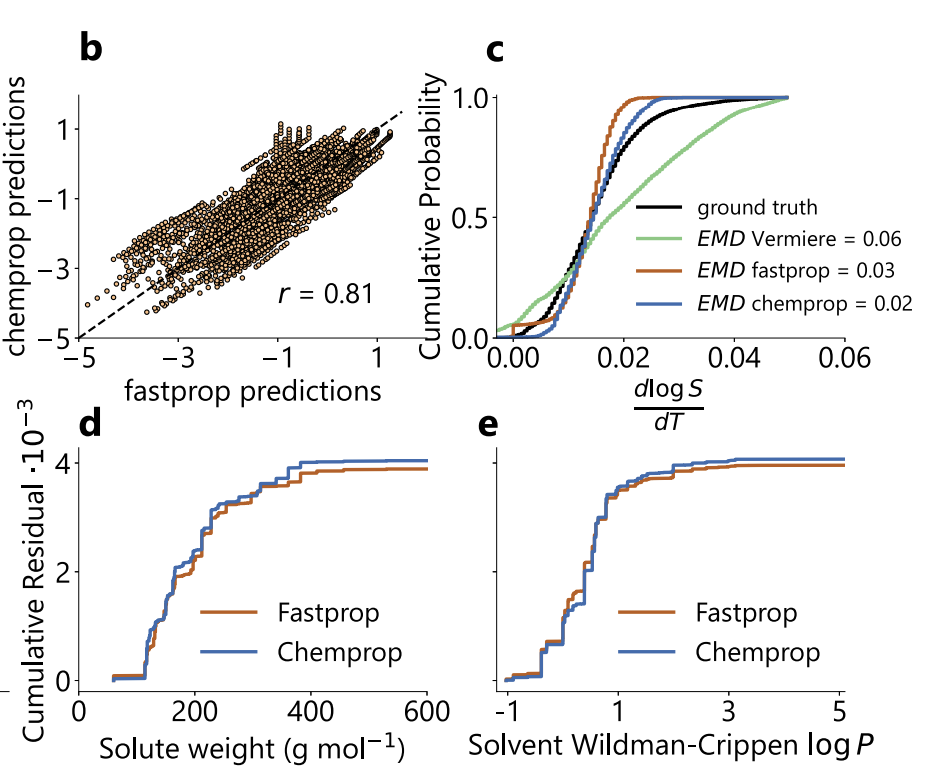
De plus, comme le montre la figure ci-dessous, les prédictions des deux modèles sont fortement corrélées sur l'ensemble de tests SolProp (r de Pearson = 0,81), et les distributions de gradient de température prédites sont également très cohérentes (EMD = 0,03/0,02). L'erreur systématique est significativement inférieure à celle du modèle de Vermeire (EMD = 0,06).
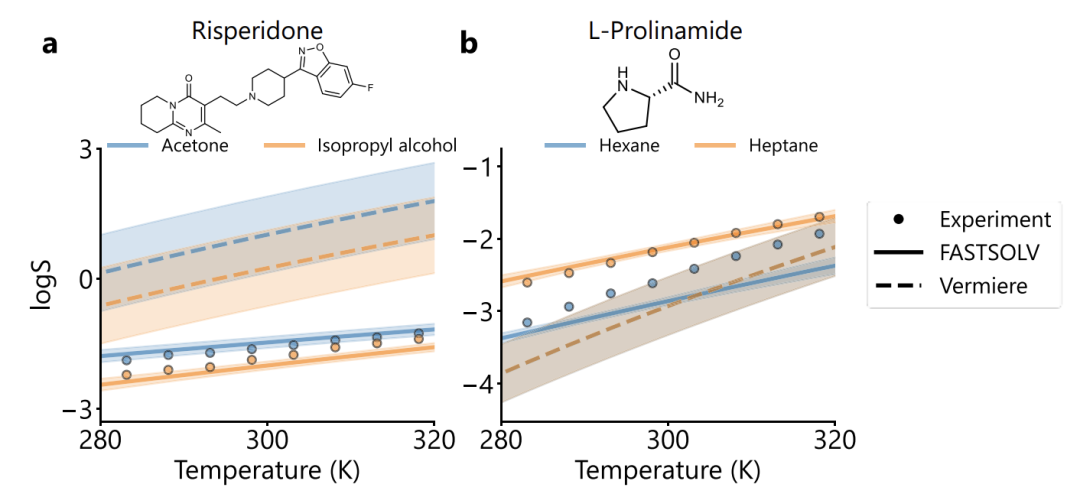
L'étude a également révélé que dans la validation typique des solutés, comme le montre la figure ci-dessous, FASTSOLV présente un avantage significatif dans la prédiction de la rispéridone (RMSE = 0,16 contre Vermeire 1,64) et de la L-proline (RMSE = 0,25 contre Vermeire 2,33).Non seulement il peut déterminer correctement l'ordre et la dépendance à la température de la solubilité du solvant, mais il peut également faire la distinction entre l'hexane et l'heptane, qui ont des structures similaires.L'analyse des modes de défaillance a montré que l'erreur de prédiction des anthraquinones était élevée, mais dans le sous-ensemble de 85 dérivés anthraquinone/anthraquinone, le RMSE global du modèle était de 0,52 et la solubilité du solvant pouvait être classée de manière stable, indiquant que la caractérisation moléculaire était raisonnable.
En résumé,Comparé au modèle Vermeire, FASTSOLV réduit le RMSE de 2 à 3 fois et accélère l'inférence jusqu'à 50 fois.Cette méthode allie interprétabilité et potentiel technique, offrant des performances de pointe dans des conditions d'extrapolation strictes. L'étude souligne également que l'ajout de données d'apprentissage supplémentaires ne dépassera pas les limites de performance, et les recherches futures se concentreront sur la constitution d'un ensemble de données de solvants organiques de haute précision.
« Dataset + IA » : une avancée mondiale dans la prédiction des propriétés moléculaires
Dans la vague actuelle d'innovation croisée en chimie, médecine et science des matériaux, la technologie de prédiction des propriétés moléculaires, centrée sur « des ensembles de données à grande échelle + des modèles d'apprentissage automatique avancés », devient un outil clé pour résoudre les problèmes de l'industrie tels que les expériences chronophages, les coûts élevés de R&D et la prédiction difficile des performances.
Dans le monde universitaire, des équipes de recherche du monde entier réagissent aux avancées de FASTSOLV et de BigSolDB en lançant une série d'études innovantes de prédiction de la solubilité. Par exemple, des chercheurs de l'Université de Leeds, au Royaume-Uni, ont proposé un modèle de relation causale structure-propriété combinant intelligence artificielle et mécanismes physico-chimiques.La prédiction de la solubilité dans un système de solvant organique et d'eau est presque aussi précise que l'erreur expérimentale.Il présente également une interprétabilité exceptionnelle et est considéré comme une étape importante dans le domaine de la modélisation de la solubilité.
Parallèlement, une équipe de recherche du Massachusetts Institute of Technology (MIT) a réalisé des progrès significatifs dans la découverte d'antibiotiques grâce au réseau neuronal graphique Chemprop. Elle a déterminé l'activité antibiotique et les profils de cytotoxicité humaine de 39 312 composés et a utilisé des ensembles de réseaux neuronaux graphiques pour prédire l'activité antibiotique et la cytotoxicité de 12 076 365 composés en vue de la découverte de nouveaux antibiotiques. En criblant un panel de composés initiaux et en évaluant leur activité inhibitrice de croissance contre la souche S. aureus RN4220 sensible à la méthicilline,512 composés actifs ont été obtenus.Le réseau neuronal graphique est ensuite formé pour effectuer des prédictions de classification binaire.
L'industrie pharmaceutique connaît également des innovations remarquables. Elle privilégie depuis longtemps les technologies d'évaluation de la solubilité à haut débit et à faible coût. Par exemple, l'outil Aspen Solubility Modeler d'AspenTech permet de prédire la solubilité de centaines de combinaisons de solvants à partir de données mesurées dans quelques solvants. Cet outil améliore considérablement l'efficacité et la fiabilité des décisions en matière de criblage de cristaux et de développement de procédés au sein de grandes entreprises comme GSK et AstraZeneca.
Par ailleurs, certaines entreprises exploitent des modèles similaires basés sur les données dans le domaine de la recherche et du développement des matériaux. En analysant de grandes quantités de données sur la structure et les performances moléculaires, elles prédisent les propriétés de nouveaux matériaux, raccourcissant ainsi les cycles de R&D et réduisant les coûts de R&D. Dans l'industrie chimique, certaines entreprises utilisent des modèles pour prédire les effets des réactions chimiques sous différents solvants et conditions de température, optimisant ainsi leurs procédés de production et améliorant leur efficacité et la qualité de leurs produits. Ce sont autant d'exemples d'entreprises appliquant des modèles et des concepts de données issus de la recherche universitaire à l'innovation industrielle concrète.
Liens de référence :
2.https://www.manufacturingchemist.com/news/article_page/Solubility_modelling/57726
Obtenez des articles de haute qualité et des articles d'interprétation approfondis dans le domaine de l'IA4S de 2023 à 2024 en un seul clic⬇️









