Command Palette
Search for a command to run...
Sur La Base De 86 000 Données De Structure De Protéines, Une Méthode D’apprentissage Automatique Combinée À Des Calculs De Mécanique Quantique a Été Utilisée Pour Découvrir 69 Nouvelles Liaisons azote-oxygène-soufre.

Dans « l’usine » cellulaire, la liaison azote-oxygène-soufre (NOS) est comme un « interrupteur intelligent » réversible qui peut réguler l’activité enzymatique en fonction des changements redox de l’environnement. En 2021, une équipe de l'Université Georg-August de Göttingen, en Allemagne, a découvert la liaison NOS entre la lysine et la cystéine en étudiant la transaldolase de Neisseria gonorrhoeae.Cette recherche va au-delà du cadre des études sur les pathogènes et les enzymes uniques et pose des bases importantes pour la science interdisciplinaire des protéines, la conception de médicaments et la bio-ingénierie.
Cependant, avec la croissance explosive des données sur la structure des protéines et la recherche continue sur les liaisons chimiques dans les structures des protéines, de nouveaux problèmes sont également apparus.Existe-t-il d’autres liaisons NOS ou interactions chimiques qui ont été négligées ?
Sur la base des considérations ci-dessus,Sophia Bazzi et Sharareh Sayyad de l'Université George Augustus ont développé un algorithme de biologie computationnelle innovant, SimplifiedBondfinder.Cela ouvre un nouveau chapitre dans l’exploration des liaisons covalentes des protéines.L'équipe a intégré l'apprentissage automatique et les calculs de mécanique quantique pour créer une base de données de cristallographie aux rayons X à haute résolution et a analysé systématiquement plus de 86 000 structures de protéines aux rayons X à haute résolution.Non seulement 69 nouvelles liaisons NOS ont été découvertes, mais elles comprenaient également de nouvelles liaisons NOS formées entre l'arginine (Arg)-cystéine et la glycine (Gly)-cystéine qui n'avaient jamais été observées auparavant.
Cette découverte révolutionnaire a élargi le champ d’application de la chimie des protéines et a rendu possible une régulation ciblée dans la conception de médicaments et l’ingénierie des protéines.Bien que cette étude se soit concentrée sur la liaison NOS, l’approche peut être appliquée de manière flexible pour étudier un large éventail d’autres liaisons chimiques et modifications covalentes.Comprend des modifications post-traductionnelles structurellement résolubles (MPT).
Les résultats de la recherche ont été publiés dans Communications Chemistry sous le titre « Révéler les liaisons arginine-cystéine et glycine-cystéine NOS par une réévaluation systématique des structures protéiques ».
Points saillants de la recherche :
* Brisant la croyance scientifique commune selon laquelle les liaisons NOS n'existent qu'entre la lysine (Lys) et la cystéine, le nouveau mécanisme de régulation redox des liaisons NOS arginine-cystéine et glycine-cystéine a été révélé pour la première fois avec une méthode innovante
* La méthode proposée intègre l'apprentissage automatique, les calculs de mécanique quantique et les données de cristallographie aux rayons X à haute résolution, résolvant le défi du manque d'algorithmes systématiques de découverte de liaisons chimiques dans ce domaine de recherche, rompant avec les limites des expériences traditionnelles et fournissant un outil fiable et facile à utiliser pour les recherches ultérieures.
* Grâce aux technologies d'apprentissage automatique et d'intelligence artificielle, le coût de ces recherches a été considérablement réduit tout en améliorant l'efficacité de la recherche, donnant l'exemple aux technologies basées sur l'apprentissage automatique pour déchiffrer les fonctions des protéines et identifier de nouvelles interactions protéiques.

Adresse du document :
https://www.nature.com/articles/s42004-025-01535-w
Autres articles sur les frontières de l'IA :
https://go.hyper.ai/UuE1o
Ensemble de données : extraction d'ensembles de données fiables avec plusieurs couches de restrictions
Les données collectées par SimplifiedBondfinder proviennent de trois bases de données de protéines différentes.Il s'agit des bases de données PDB, PDB-REDO et BDB. Les données collectées seront soumises à diverses contraintes afin de filtrer des ensembles de données fiables et exploitables. Parmi elles, la base de données PDB-REDO (version de janvier 2024) affine et optimise la structure statique de la base de données PDB afin de la rendre plus conforme aux normes cristallographiques actuelles. Comparée à l'entrée PDB originale, elle offre une précision et une fiabilité supérieures. Comme illustré à gauche de la figure ci-dessous :
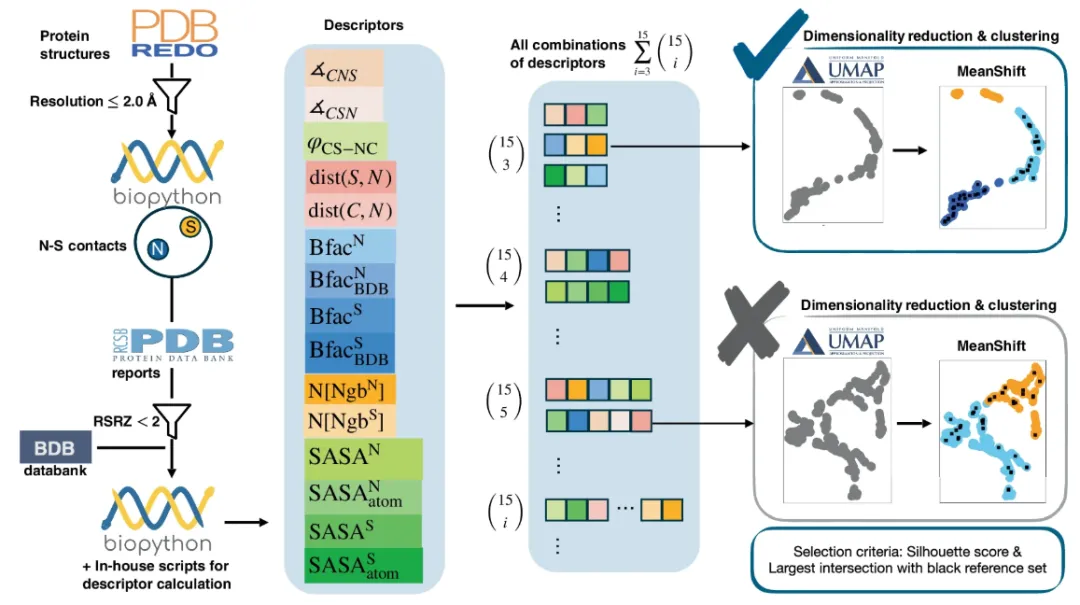
Plus précisément, l’équipe de recherche a utilisé plusieurs fonctions interdépendantes pour piloter la génération automatique d’ensembles de données dans une base de données qui contenait initialement 170 251 données sur les protéines.L'équipe de recherche a d'abord utilisé Biopython (v 1.79) pour réaliser des analyses structurales (avec MMCIFParser et PDBParse) et calculer d'autres propriétés atomiques et résiduelles. Après avoir analysé uniquement la structure déterminée par rayons X, l'équipe de recherche a optimisé les données de 170 127 protéines.
Par la suite, afin d’améliorer encore la précision de la prédiction, l’équipe de recherche a examiné davantage les structures protéiques avec une résolution de ≤ 2 Å et a finalement obtenu 86 491 structures pour une analyse expérimentale.
Pour construire un ensemble de données permettant d'étudier une liaison chimique spécifique,L’équipe de recherche a établi des critères basés sur les types d’atomes constitutifs, les noms de résidus, les distances interatomiques et l’occupation.Pour les connexions NOS impliquant des atomes de soufre (S) et d'azote (N) dans les résidus standards, l'équipe de recherche a limité la distance interatomique de SN, c'est-à-dire dist(S, N), à ≤ 3,2 Å, correspondant à la valeur limite des interactions de valence entre la lysine et la cystéine, et a fixé le seuil d'occupation à > 0,8 pour exclure les atomes présentant une forte incertitude de position. Grâce à ce critère, l'étude a identifié 25 462 contacts NS.
Pour garantir la représentation de la masse atomique cible, l'équipe de recherche a en outre appliqué le score Z de la valeur R de l'espace réel (RSRZ) avec un seuil défini à < 2,0 pour garantir que des correspondances fiables avec les données dans l'espace réel puissent être identifiées.L'ensemble de données a été réduit à 23 129 contacts NS.Cela a permis aux cibles expérimentales de se concentrer principalement sur deux types d'interactions de la cystéine : l'interaction entre l'atome de soufre de la cystéine et l'azote de la chaîne principale de la glycine ; et l'interaction entre l'atome de soufre de la cystéine et l'azote de la chaîne latérale de l'arginine et de la lysine.
Suivant,L'équipe de recherche a utilisé le module NeighborSearch de Biopython pour extraire des paramètres structurels, en collectant 15 descripteurs différents pour chaque échantillon dans chaque ensemble de données.Il s'agit notamment des angles (∡CSN, ∡CNS), des angles de torsion (φCS-NC), d'autres distances (dist(C, N), dist(S, N)) et des valeurs de surface accessible au solvant (SASA) des atomes cibles et des résidus correspondants calculés ultérieurement à l'aide de Bio.PDB.SASA.
L'équipe de recherche a inclus des facteurs B atomiques (Bfac) dans l'expérience afin d'obtenir un paramètre de mobilité atomique cible dans l'analyse. Ces valeurs proviennent de deux bases de données : la base de données PDB du RCSB et une base de données de fichiers PDB (BDB) contenant des facteurs B cohérents.
Il convient de mentionner que, sur la base des exigences spécifiques de cette étude, seuls 15 descripteurs ont été sélectionnés dans l’expérience.Cependant, l’équipe de recherche a déclaré que l’algorithme proposé n’a pas de limite stricte quant au nombre de descripteurs qu’il peut traiter.De par sa conception, il peut accueillir un nombre arbitraire de descripteurs, ce qui lui permet d'intégrer des connaissances spécifiques à un domaine ou de s'adapter à de nouvelles approches expérimentales.
Architecture du modèle : intégration de l'apprentissage automatique et du calcul de la mécanique quantique
La partie ci-dessus est la première étape des étapes clés de la méthode proposée, qui consiste à construire un ensemble de données cibles pour des liaisons chimiques spécifiques et à appliquer des critères stricts.Cette section se concentre sur la deuxième étape clé de la méthode proposée, qui consiste à utiliser des techniques d’apprentissage automatique pour explorer ces données de grande dimension.Identifier les descripteurs structurels efficaces et prédire les sites potentiels de formation de liaisons covalentes.
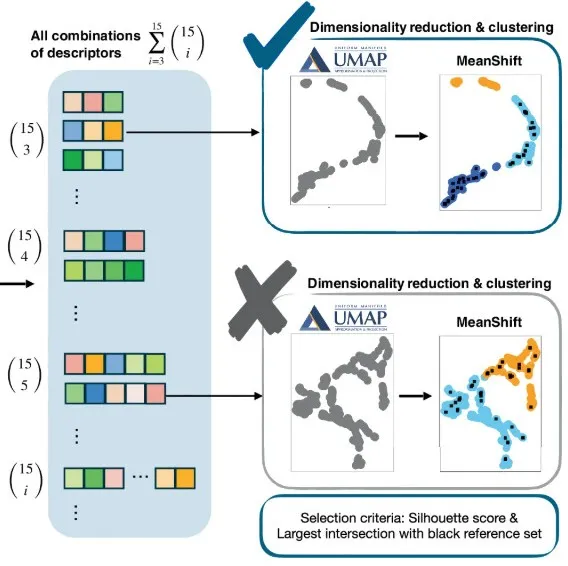
Comme le montre l'image ci-dessus.Dans un premier temps, l’équipe de recherche a appliqué la technique de réduction de dimensionnalité non supervisée par approximation et projection uniformes de collecteurs (UMAP) avec une dimension d’intégration maximale de 3.Ensuite, un regroupement par décalage moyen est effectué sur tous les ensembles de descripteurs possibles.
dans,UMAP préserve de manière optimale les propriétés topologiques et géométriques intrinsèques des données de grande dimension, garantissant que les caractéristiques structurelles essentielles sont préservées dans les intégrations de faible dimension.Cela facilite une analyse en aval pertinente. Le choix de la dimension d'intégration dans UMAP dépend des propriétés topologiques et géométriques de l'ensemble de données et de sa variété d'origine à haute dimension. Dans les applications pratiques, les intégrations bidimensionnelles ou tridimensionnelles sont les plus interprétables, car elles permettent une visualisation et une évaluation intuitives de la qualité du clustering.
Dans cette étude, trois dimensions d'intégration ont fourni des clusters bien séparés et significatifs, justifiant ce choix. L'analyse des liaisons chimiques et les résultats du clustering montrent que cette méthode de réduction de dimensionnalité est optimale pour l'ensemble de données utilisé dans cette expérience. Sélectionner des dimensions d'intégration supérieures à celles nécessaires permet de préserver les caractéristiques originales populaires, mais augmente les coûts de calcul sans améliorer l'interprétabilité. À l'inverse, réduire la dimensionnalité en dessous du niveau optimal entraînera une perte d'information importante et une mauvaise séparation des clusters.
Alors,L'équipe de recherche a obtenu le score Silhouette de toutes les coordonnées d'intégration tridimensionnelles pour évaluer la qualité de regroupement de chaque combinaison.L'algorithme génère des clusters, des coefficients de silhouette et la connectivité référence-cible au sein de chaque cluster. Chaque candidat est identifié par le nom de l'atome cible, le nom du résidu correspondant, le numéro du résidu, la chaîne et l'identifiant PDB afin de distinguer tous les atomes cibles de la protéine.
Pour trouver l’espace de caractéristiques final et minimal, l’équipe de recherche a utilisé plusieurs critères, notamment la valeur du coefficient de silhouette, le nombre de clusters produits par chaque espace de caractéristiques et la distribution des connexions référence-cible dans ces clusters.
Spécifiquement,L’équipe de recherche a cherché à identifier un espace de caractéristiques capable de segmenter efficacement les données en deux ou trois groupes distincts avec un coefficient de silhouette ≥ 0,5.Idéalement, l'un des clusters ne contient aucune connexion cible de référence, ce qui est appelé « cluster impossible ». En pratique, le nombre minimal d'échantillons de référence dans ce cluster est acceptable. Les clusters restants, contenant la totalité ou la plupart des connexions cibles de référence, sont appelés « clusters possibles ».
En introduisant des clusters candidats possibles et impossibles contenant les liaisons chimiques cibles,L’équipe de recherche a pu identifier des espaces de caractéristiques optimisés pour distinguer les paires d’atomes cibles susceptibles de former de nouvelles liaisons chimiques et celles qui sont moins susceptibles de former de telles liaisons.Une fois qu'un ensemble de descripteurs permettant de distinguer ces cas de manière fiable est identifié, il n'est plus nécessaire d'inclure des descripteurs supplémentaires. Cette approche présente des avantages en termes d'efficacité de calcul et d'interprétabilité, et pourrait améliorer considérablement la précision prédictive des méthodes d'identification de la formation de nouvelles liaisons chimiques au sein des structures protéiques.
En plus de l’apprentissage automatique, la méthode proposée dans cette étude intègre également des calculs de mécanique quantique.Français Les chercheurs ont optimisé la géométrie des candidats potentiels pour la liaison NOS dans les complexes Lys-NOS-Cys, Gly-NOS-Cys, ARG-NηOS-Cys et ARG-NεOS-Cys. L'optimisation géométrique a été réalisée dans l'eau à l'aide du progiciel Gaussian16 – A.03 (Gaussian 16, révision C.01) au niveau théorique B3LYP-D3 (BJ)/def2-TZVPD. Pour les structures optimisées, plusieurs paramètres géométriques ont été calculés expérimentalement, notamment la distance entre les atomes de soufre et d'azote (dist (S, N)), ainsi que les angles (∡CSN, ∡CNS, ∡NOS).
Afin de vérifier l'existence de la liaison covalente NOS prédite par la méthode de clustering proposée,L'équipe de recherche a utilisé phenix.refine (version 1.20.1-4487-000) pour réoptimiser quatre structures protéiques représentatives ;Une validation structurale complète a été réalisée à l'aide de phenix.molprobity afin d'évaluer la qualité géométrique, les scores de collision et les interactions stériques, afin de garantir la cohérence avec les données cristallographiques haute résolution. Un rapport de validation complet a été généré à l'aide de phenix.table1, résumant les statistiques de raffinement, les indicateurs de qualité du modèle et les écarts stéréochimiques. Ces étapes de validation ont confirmé l'intégrité structurale de la jonction NOS et sa compatibilité avec la carte de densité électronique.
Résultats expérimentaux : les liaisons Arg-NOS-Cys et Gly-NOS-Cys sont des liaisons covalentes raisonnables
Pour démontrer l’efficacité de la méthode proposée, l’équipe de recherche a mené un certain nombre d’expériences, explorant l’utilisation de techniques d’apprentissage automatique pour la sélection de descripteurs, la signification biochimique de l’espace multi-descripteurs, l’analyse de clusters et la vérification structurelle et thermodynamique.
Sélection de descripteurs à l'aide de l'apprentissage automatique
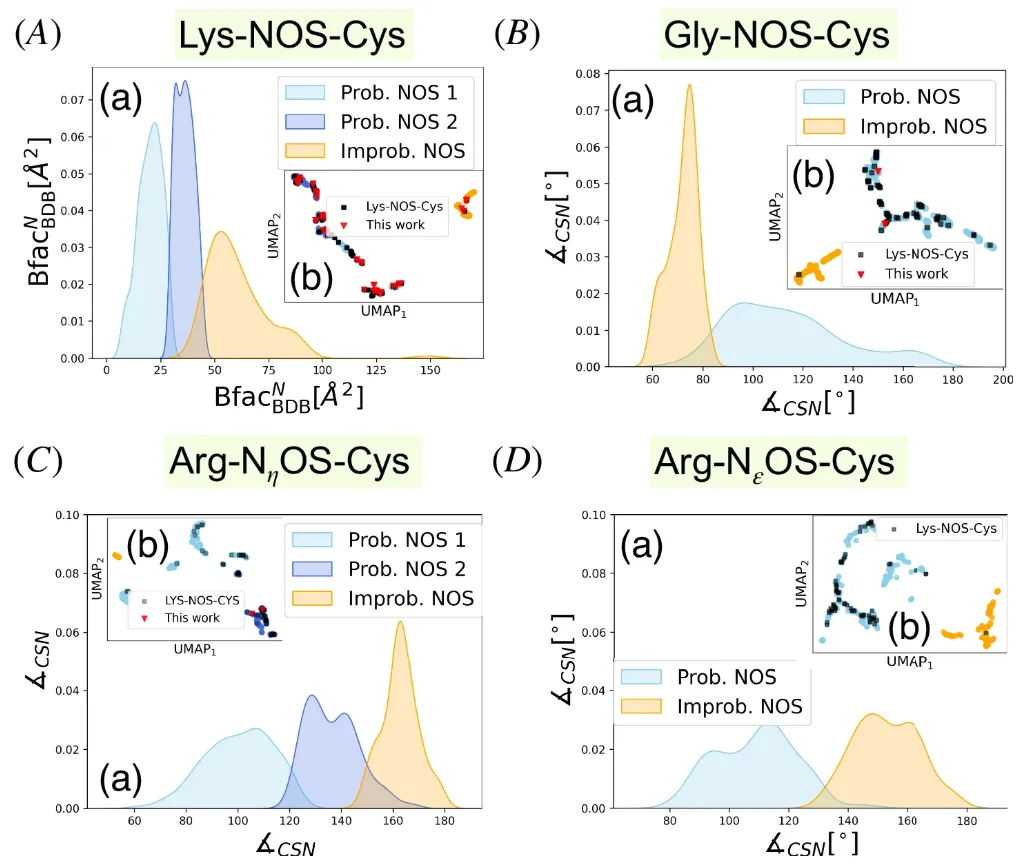
L’équipe de recherche l’a d’abord appliqué à des données où des connexions Lys-NOS-Cys étaient susceptibles d’exister.L'ensemble de données contient 527 paires lysine-cystéine et comprend également des liaisons NOS vérifiées expérimentalement.Les descripteurs clés ont été déterminés expérimentalement comme étant le facteur B de l'atome d'azote (Bfac(BDB)(N)) et le nombre de résidus voisins dans un rayon de 4 Å des atomes Cα de la lysine (Ngbᴺ) et de la cystéine (Ngbˢ).
L'équipe de recherche a ensuite étendu l'analyse à un ensemble de données de 313 paires glycine-cystéine pour explorer les connexions potentielles Gly-NOS-Cys, comme le montre la figure ci-dessous.
Ici, les ensembles de descripteurs clés incluent le facteur B des résidus contenant du soufre (BfacBDBS), la distance soufre-azote (dist(S,N)) et l'angle carbone-soufre-azote (∡CSN).
En termes de descripteurs clés pour prédire la formation de liaisons NOS entre les résidus d'arginine et de cystéine,La chaîne latérale de l'arginine possède deux types d'atomes d'azote, Nη et Nε, qui diffèrent par leurs caractéristiques géométriques et leurs propriétés chimiques.Par conséquent, nous avons analysé les ensembles de données Nη (Arg-NηOS-Cys) et Nε (Arg-Nε-Cys) séparément.
Pour Arg-NηOS-Cys, les descripteurs sélectionnés correspondent à la surface accessible au solvant du résidu d'azote (SASAᴺ), ∡CSN et aux résidus adjacents au soufre (Ngbˢ) et à l'azote (Ngbᴺ) ; de même pour l'ensemble de données de 240 paires Arg-NεOS-Cys, les descripteurs clés impliquent BfacBDBS, SASAˢ, la surface accessible au solvant de l'atome d'azote, ∡CSN et ∡CNS.
Ces résultats montrent une séparation claire des clusters grâce à la visualisation de la réduction de la dimensionnalité UMAP.Comme le montre la figure ci-dessous, le bleu ciel et le bleu roi représentent les candidats à la liaison NOS, l'orange représente le « clustering impossible » et les carrés noirs représentent l'ensemble de données de référence. On constate clairement que la distribution des échantillons susceptibles de former des liaisons NOS se superpose fortement à celle des points de référence.
Signification biochimique de l'espace descripteur multidimensionnel
L'équipe de recherche a exploré la pertinence biochimique des descripteurs clés, qui étaient importants pour distinguer les liaisons NOS des liaisons non NOS, en déterminant algorithmiquement un ensemble minimal de descripteurs.
Prenons l'exemple du facteur B : sa distribution varie selon le groupe. Comme le montre la figure A(a) ci-dessus, les modes de distribution du facteur B diffèrent selon qu'il s'agit d'un « groupement possible » ou d'un « groupement impossible ».Le facteur B est lié à la flexibilité des atomes ou des régions, et les résidus du site actif ont généralement un facteur B inférieur, indiquant qu'ils sont liés à l'activité enzymatique.Cependant, l’équipe de recherche a également souligné que le faible facteur B peut indiquer une liaison NOS, mais il peut également refléter d’autres interactions azote-soufre.
En ce qui concerne les caractéristiques descriptives des liaisons NOS formées par différents résidus d'acides aminés, BfacBDBᴺ est le principal facteur distinguant les deux clusters dans Lys-NOS-Cys ; pour les connexions Gly-NOS-Cys, ∠CSN est le principal descripteur distinguant les clusters de connexions NOS possibles, avec ∠CSN > 80° pour la plupart des échantillons possibles et la valeur ∠CSN du complexe Gly-NOS-Cys optimisé étant d'environ 94° ; ∠CSN est toujours le déterminant clé pour distinguer les connexions NOS possibles des connexions impossibles pour les connexions Arg-NεOS-Cys.
Analyse de cluster
Dans cette évaluation, l’équipe de recherche a détecté 65 liaisons Lys-NOS-Cys, 2 liaisons Gly-NOS-Cys (Figures a et b ci-dessous) et 2 liaisons Arg-NηOS-Cys (Figures c et d ci-dessous).
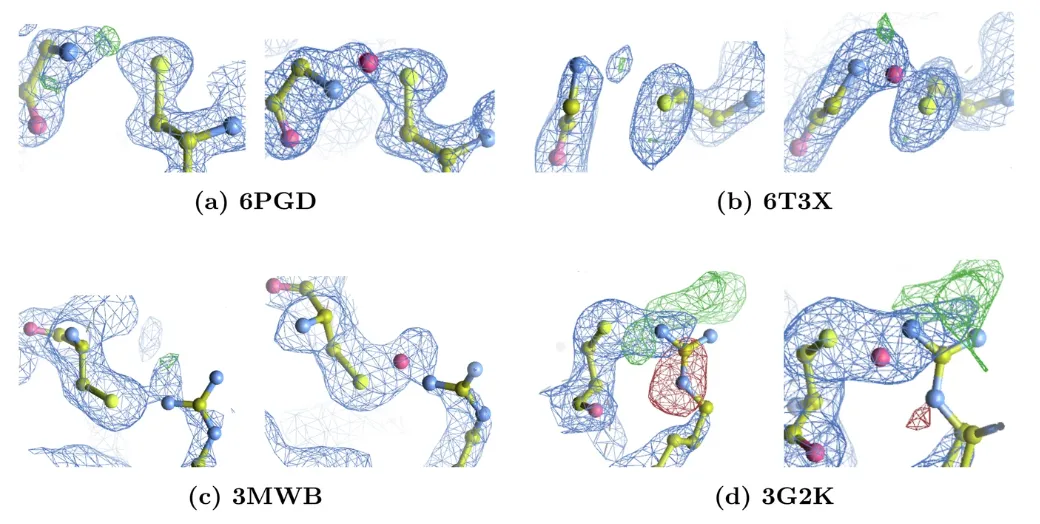
Grâce à une modélisation explicite et à un ré-affinement, l'équipe de rechercheAprès l'introduction des liaisons NOS, la valeur Rwork/Rfree s'est améliorée en moyenne de 0,5% et le pic de densité électronique inexpliqué a été considérablement réduit.Pour 3G2K, on observe un pic de densité électronique négatif autour de la chaîne latérale d'arginine dans la structure d'origine, qui est significativement réduit après la redistribution de la conformation d'arginine. De plus, on observe des pics de différence positifs près de la chaîne latérale d'arginine dans les deux modèles. En raison de leur grande amplitude et de la présence de DMSO, ils pourraient représenter des molécules de solvant non modélisées dans le modèle actuel.
Vérification structurelle et thermodynamique
Pour confirmer davantage le lien entre Arg-NOS-Cys et Gly-NOS-Cys, l'équipe de recherche a combiné l'optimisation de la géométrie mécanique quantique avec l'évaluation thermodynamique de quatre complexes protéiques représentatifs (6PGD, 6T3X, 3MWB et 3G2K) pour expliquer systématiquement la possible variabilité chimique in vivo.
En termes de vérification structurelle, dans le modèle optimisé de liaison NOS, la distance SN varie de 2,61 à 2,70 Å, ce qui est très proche de l'intervalle de 2,63 à 2,89 Å de la structure PDB-REDO d'origine.Les simulations avec l'atome d'oxygène de pontage supprimé ont entraîné une augmentation significative de la séparation SN à 3,36-4,26 Å, indiquant que les distances SN plus courtes observées expérimentalement sont cohérentes avec la présence d'un atome d'oxygène intermédiaire.
En termes d'évaluation thermodynamique, l'équipe de recherche a calculé l'énergie libre de Gibbs (ΔG) sous différents états de protonation, montrant que tous les processus de formation de liaisons NOS sont négatifs.Cela suggère que le remplacement d'un hydrogène par un oxygène pour former la liaison NOS est thermodynamiquement réalisable dans l'état simulé. Cependant, l'amplitude de ΔG diffère significativement selon l'état de protonation et entre les complexes dérivés de l'arginine et de la glycine. Dans les deux systèmes, la glycine neutre ou l'arginine est favorisée par rapport aux états chargés positivement. Les complexes à base de glycine présentent des valeurs de ΔG légèrement plus élevées. Bien que ces valeurs impliquent toujours une association thermodynamiquement favorable, elles sont systématiquement moins exergoniques que les complexes d'arginine correspondants.
Pris ensemble, ces résultats structurels fournissent des preuves cohérentes queIl est démontré que les liaisons Arg-NOS-Cys et Gly-NOS-Cys sont des liaisons covalentes raisonnables plutôt que de simples contacts non liés.Dans le même temps, l'accord entre les géométries optimisées mécaniquement quantique et les données cristallographiques du système cristallin, ainsi que les énergies libres négatives de formation, suggèrent fortement que ces connexions sont structurellement et énergétiquement réalisables dans l'environnement protéique concerné.
L'apprentissage automatique ouvre un nouveau chapitre dans le monde microscopique des protéines
Comme mentionné dans l'article, le développement rapide des technologies d'apprentissage automatique et d'intelligence artificielle a démontré leur supériorité sur les méthodes biochimiques traditionnelles pour résoudre des problèmes biochimiques complexes. Grâce à leur faible coût de calcul et à leur grande efficacité, ces technologies ont incité la communauté scientifique à révolutionner les méthodes de production et ont également permis aux technologies basées sur l'apprentissage automatique d'exploiter pleinement leur potentiel pour déchiffrer les fonctions des protéines et identifier de nouvelles interactions.
Par coïncidence, Kevin K. Yang et al. du California Institute of Technology ont publié un article intitulé « Machine learning-guided directed evolution for protein engineering » dans Nat. Methods.En comparant l’évolution dirigée et l’évolution dirigée assistée par l’apprentissage automatique, la supériorité de l’apprentissage automatique est expliquée.Dans le même temps, l'article répertorie également des cas pratiques tels que l'efficacité catalytique des enzymes et l'optimisation de la stabilité thermique du cytochrome P450, et mentionne une variété de méthodes d'apprentissage automatique telles que la régression linéaire, le processus gaussien et l'optimisation bayésienne.Cela montre que l’apprentissage automatique peut fournir une « navigation intelligente basée sur les données » pour l’ingénierie des protéines.En modélisant les relations séquence-fonction, l’efficacité et le taux de réussite de l’évolution dirigée peuvent être considérablement améliorés.
Adresse du document :
https://arxiv.org/pdf/1811.10775
En outre, un article publié par Rita Casadio et al. de l'Université de Bologne en Italie intitulé « Solutions d'apprentissage automatique pour prédire les interactions protéine-protéine » détaille également l'exploration de l'apprentissage automatique dans la recherche sur les protéines.Il présente l'application de méthodes d'apprentissage automatique, notamment l'apprentissage non supervisé et supervisé dans les interactions moléculaires protéine-protéine (PPI).Les principaux problèmes liés à la qualité des données, à la représentation, aux algorithmes de formation et aux procédures de validation sont mis en évidence.
Adresse du document :
https://wires.onlinelibrary.wiley.com/doi/full/10.1002/wcms.1618
En général, il existe encore de nombreux codes liés à la vie cachés dans le monde microscopique des protéines, et la méthode systématique basée sur les données avec l'apprentissage automatique comme moyen principal est sans aucun doute comme une clé pour ouvrir la porte au monde microscopique des protéines, inspirant la communauté de recherche scientifique à mener des recherches et des explorations plus approfondies de la fonction et de la stabilité des protéines, brisant ainsi constamment les limites de la cognition humaine de la vie.








