Command Palette
Search for a command to run...
Sélectionné Pour l'ICML 2025, l'Université Tsinghua/Université Renmin De Chine/ByteDance a Proposé Le Premier Cadre De Génération Unifiée Intermolécule UniMoMo Pour Réaliser La Conception De Molécules Médicamenteuses Multitypes

Le groupe dirigé par le professeur Liu Yang de l'Université Tsinghua, le groupe dirigé par le professeur Huang Wenbing de la Gaoling School of Artificial Intelligence de l'Université Renmin de Chine et l'équipe pharmaceutique ByteDance AI ont proposé conjointement un cadre de génération unifié à travers les espèces moléculaires, UniMoMo.Ce cadre représente uniformément différents types de molécules sur la base de fragments moléculaires (blocs), utilise des autoencodeurs variationnels pour compresser la conformation atomique complète de chaque bloc et effectue une modélisation de diffusion géométrique dans l'espace latent compressé, permettant ainsi la conception de différents types de molécules de liaison (petites molécules, peptides, anticorps) pour la même cible. UniMoMo atteint des performances de premier plan dans de multiples tests de tâches moléculaires, démontrant le grand potentiel du transfert de connaissances intermodal et du partage de données.

Les résultats associés ont été sélectionnés pour ICML 2025 sous le titre « UniMoMo : Modélisation générative unifiée de molécules 3D pour la conception de liants de novo ».
Adresse du document :
Adresse du projet open source :
https://github.com/kxz18/UniMoMo
Pourquoi la modélisation unifiée ?
Différents types moléculaires ont leurs propres avantages et inconvénients dans le développement de médicaments, il est donc souvent nécessaire de sélectionner le type moléculaire le plus approprié dans différents scénarios de maladie. Par exemple:
* Les petites molécules sont de petite taille, faciles à prendre par voie orale et ont une forte pénétration, ce qui les rend adaptées pour pénétrer dans les cellules et agir sur les cibles. Ils sont largement utilisés dans les maladies chroniques et les maladies métaboliques ;
* Les molécules peptidiques ont des propriétés de ciblage élevées et peuvent se lier à des zones larges et plates à la surface des protéines. Ils sont adaptés pour cibler les sites d'interaction protéique « difficiles à traiter » et sont souvent utilisés dans le traitement du cancer, de l'inflammation, etc.
* Les anticorps ont une sélectivité et une affinité extrêmement élevées et peuvent identifier de manière stable des marqueurs protéiques spécifiques, ce qui les rend particulièrement adaptés à des scénarios d'intervention précis tels que l'immunothérapie.
Par conséquent, face à des mécanismes pathologiques, des caractéristiques cibles et des besoins médicamenteux différents, les types de molécules adaptées à l’utilisation sont différents. Les méthodes génératives existantes ne modélisent généralement qu’une certaine classe de molécules (telles que les petites molécules, les peptides ou les anticorps).Il ne peut ni répondre à des besoins thérapeutiques divers ni utiliser les points communs entre différentes molécules pour améliorer les performances du modèle.
D'un point de vue applicatif, la modélisation unifiée nous permet d'explorer simultanément plusieurs types de candidats médicaments pour la même cible, offrant ainsi davantage d'options pour différents scénarios en aval.
Du point de vue de l'apprentissage automatique, différents types de molécules partagent des règles de liaison similaires (liaisons hydrogène, empilement π-π, ponts salins, etc.) et des contraintes géométriques (longueurs de liaison, angles de liaison, etc.), et peuvent apprendre les unes des autres.Par conséquent, la modélisation unifiée devrait être en mesure d’améliorer les capacités de généralisation et de transfert croisé du modèle en utilisant une échelle de données plus grande.
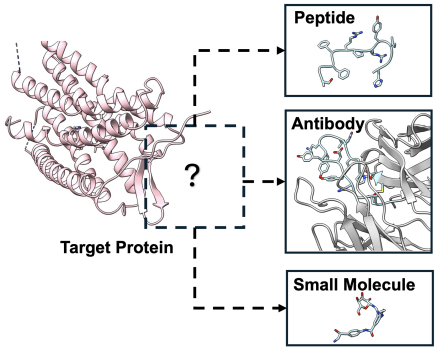
La difficulté de la modélisation générative unifiée
Bien que l’idée de générer uniformément différents types de molécules soit passionnante, il existe encore d’énormes défis dans la réalisation d’un tel cadre, principalement dans le choix de la représentation moléculaire et la conception de l’algorithme de génération.
Premièrement, il existe de grandes différences dans la représentation structurelle des différents types moléculaires : les petites molécules sont composées de divers groupes fonctionnels et leurs structures sont très diverses et non linéaires ; tandis que les peptides et les anticorps sont composés d'acides aminés connectés dans une séquence linéaire, et les anticorps en particulier ont des divisions claires des régions fonctionnelles. Une approche intuitive mais peu efficace consiste à modéliser toutes les molécules sous forme de graphiques d’atomes.Cependant, cette approche ignore la structure hiérarchique naturelle des molécules, comme les sous-structures clés telles que les cycles benzéniques ou les acides aminés standard, et conduit à des coûts de calcul extrêmement élevés lorsqu'il s'agit de systèmes avec de grandes surfaces de liaison comme les anticorps.
Au contraire, si seuls des vocabulaires de fragments structurels communs sont utilisés pour construire des graphes au niveau des fragments (par exemple, la plupart des travaux de conception de protéines ne prennent en compte que Cα coordonner),Ignorer les détails au niveau atomique sacrifiera la portabilité et la précision de la génération de molécules.Étant donné que les lois essentielles de la conception des molécules de liaison sont l'interaction spatiale avec la cible et les contraintes géométriques au sein de la molécule, il s'agit de lois physiques définies au niveau atomique et nécessitant un support d'information précis sur tous les atomes.
Par conséquent, pour construire une représentation moléculaire unifiée véritablement efficace et efficiente, deux défis doivent être résolus simultanément :Il est nécessaire de conserver les détails géométriques au niveau atomique tout en faisant abstraction des priors hiérarchiques structurels.
Deuxièmement, si des fragments structurels sont introduits dans la génération pour préserver la priorité hiérarchique, cela posera des défis majeurs à l'algorithme de génération :Les modèles de diffusion traditionnels s’appuient généralement sur des représentations de données à longueur et structure fixes.Par exemple, un nombre fixe de nuages de points ou d’atomes. Pour les modèles de prédiction de structure tels que AF3, étant donné que la topologie 2D est donnée à l'avance, le processus de diffusion n'entraînera pas de changements dans le nombre d'atomes ou dans la structure 2D. Pour la tâche de génération moléculaire, la topologie 2D et la structure 3D doivent être générées simultanément. Lorsque le type de fragments structurels change au cours du processus de débruitage, le nombre, le type et la disposition des atomes correspondants changeront également en conséquence. Cela brise les hypothèses des modèles de diffusion conventionnels et impose des exigences extrêmement élevées en matière de modélisation.
UniMoMo : un modèle génératif unifié
Afin de résoudre le problème des grandes différences structurelles et de la grande difficulté de modélisation des différents types moléculaires, l'article propose un nouveau cadre - UniMoMo.Cela commence par deux conceptions clés, prenant efficacement en compte la hiérarchie structurelle et la précision au niveau atomique :
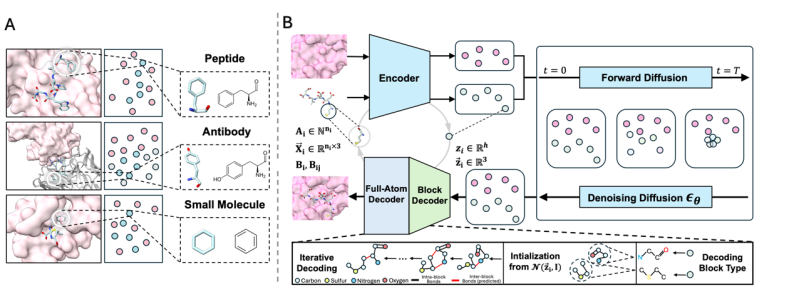
* Représentation unifiée :Tous les types de molécules sont modélisés sous forme de blocs.
Qu'il s'agisse d'une petite molécule, d'un peptide ou d'un anticorps, UniMoMo représente sa structure sous la forme d'un graphique composé de fragments moléculaires (blocs). Chaque bloc peut être un acide aminé standard ou un fragment de petite molécule commun (tel qu'un cycle benzénique, un indole, etc.). Dans la mise en œuvre de l'article, les fragments moléculaires enregistrés incluent tous les acides aminés standard et les fragments de petites molécules automatiquement identifiés par l'algorithme d'exploration de sous-graphes principaux. Tous les acides aminés non naturels peuvent être classés comme de petites molécules.Cette représentation conserve à la fois les détails au niveau atomique des molécules et la structure hiérarchique des différents types de molécules elles-mêmes, rendant possible une modélisation unifiée.
* Modèle de diffusion spatiale implicite géométrique tout atome :Génération efficace sur des représentations compressées.
Afin de résoudre le problème des changements synchrones dans le type et la quantité d'atomes causés par les changements dans les types de blocs pendant le processus de génération, et d'améliorer l'efficacité de la génération et la précision structurelle,L'article conçoit un autoencodeur variationnel itératif entièrement atomique (IterVAE).Tous les atomes de chaque bloc sont compressés en un « point » dans l'espace latent, comprenant un vecteur de représentation de l'espace latent de longueur fixe et les coordonnées de l'espace latent correspondantes.
Le modèle effectue ensuite une modélisation générative dans cet espace latent géométrique compressé pour générer des représentations latentes de nouvelles molécules, qui sont finalement décodées jusqu'à la structure atomique complète.Étant donné que la représentation des données de l'espace latent est de longueur fixe (le nombre de blocs est prédéfini) et continue, elle peut être facilement compatible avec divers algorithmes de génération existants.Dans les tentatives actuelles, les modèles de diffusion ont pu produire des résultats relativement bons. Cette conception permet au modèle de se concentrer sur la disposition globale entre les blocs pendant le processus de génération, tandis que la structure détaillée au niveau atomique est complétée par le décodeur, réalisant ainsi l'unité d'une efficacité élevée et d'une précision au niveau atomique.
La modélisation unifiée va au-delà de la modélisation à domaine unique
Afin de vérifier la polyvalence et l’efficacité d’UniMoMo sur différents types de molécules, les auteurs ont mené une évaluation systématique dans plusieurs tâches de conception basées sur la structure.Il couvre trois types représentatifs de molécules de liaison : les petites molécules, les peptides et les anticorps.En comparant avec le modèle de génération de type de molécule unique le plus représentatif dans le domaine correspondant, l'expérience vise à explorer si la modélisation unifiée a des capacités de modélisation géométrique et de généralisation intermodale plus fortes, notamment en termes d'indicateurs clés tels que la rationalité de la structure spatiale et la capacité de liaison.
Les résultats montrent queUniMoMo, formé de manière uniforme, a atteint une supériorité globale dans tous les types moléculaires.Non seulement il excelle dans la précision de la restauration structurelle, mais il permet également d’obtenir des améliorations significatives dans la rationalité géométrique clé et la qualité de l’interaction avec la cible.
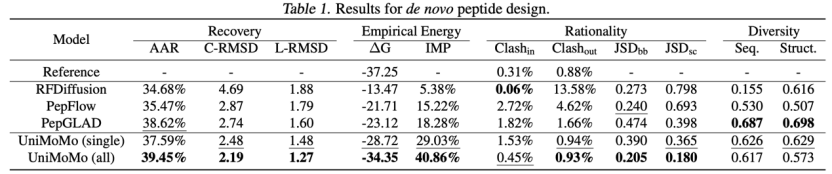
Dans la tâche de génération de peptides,UniMoMo surpasse considérablement les modèles spécifiques au domaine existants dans plusieurs indicateurs clés.Y compris RFDiffusion, PepFlow et PepGLAD, etc. En particulier en termes de précision structurelle, UniMoMo a obtenu un RMSD inférieur du complexe et du monomère, indiquant que les structures peptidiques qu'il a générées étaient plus proches de la conformation de liaison réelle.
UniMoMo peut également générer des structures avec des énergies de liaison Rosetta inférieures.Cela reflète sa plus grande capacité de modélisation des caractéristiques géométriques des sites de liaison des protéines.En outre, UniMoMo a également montré des performances de premier plan dans les indicateurs de rationalité géométrique tels que la cohérence de la distribution des angles dièdres (JSD des torsions de la chaîne principale/latérale) et le conflit spatial au niveau atomique (taux de conflit) qui mesurent la qualité de la conformation des peptides. De plus, UniMoMo (tous), qui a été formé en utilisant toutes les données, a systématiquement surpassé le modèle formé en utilisant uniquement des données peptidiques dans divers indicateurs.La capacité d’UniMoMo à apprendre et à généraliser à travers les espèces moléculaires est démontrée.
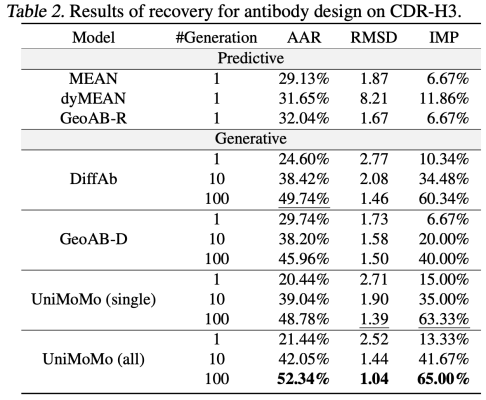
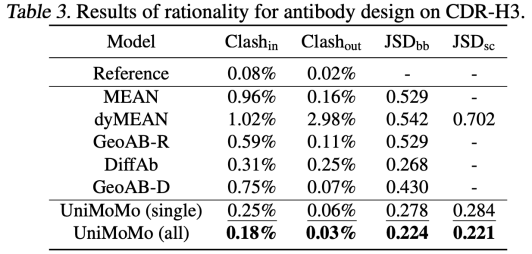
UniMoMo a également démontré de solides performances dans la tâche de conception d'anticorps. Par rapport aux méthodes existantes telles que MEAN, dyMEAN et DiffAb,UniMoMo surpasse tous les autres objectifs en termes d'indicateurs clés tels que le rappel des séquences et structures naturellement liées (AAR et RMSD) et l'amélioration de l'énergie de liaison (IMP).En particulier dans l'évaluation de la génération d'échantillonnages multiples, UniMoMo est capable de générer des fragments d'anticorps proches de la conformation naturelle avec une probabilité plus élevée, montrant sa bonne capacité d'exploration dans l'espace de structure des anticorps.
De même, UniMoMo(all), qui est formé conjointement à l’aide de données provenant de différents types moléculaires, surpasse la version formée uniquement à l’aide de données d’anticorps dans tous les indicateurs.Cela montre que la modélisation unifiée aide le modèle à apprendre des lois spatiales plus universelles et transférables des structures moléculaires.Ce résultat met en évidence les points communs dans la modélisation structurelle entre différents types moléculaires et vérifie la valeur significative de la fusion de données inter-domaines pour améliorer la qualité de la génération.
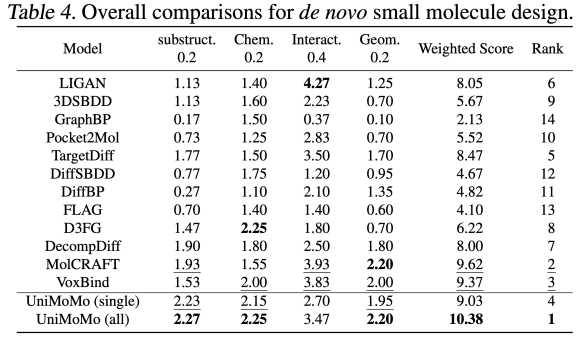
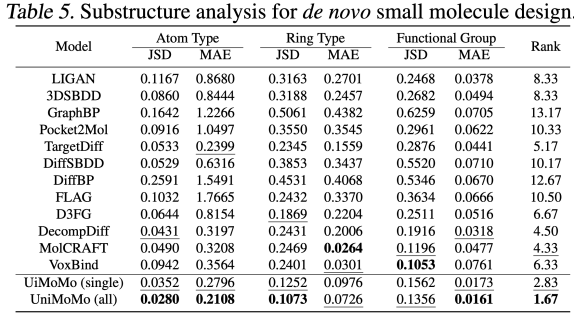
UniMoMo a également démontré des performances supérieures dans les tâches de génération de petites molécules. Grâce à l'évaluation de l'ensemble de données CrossDocked2020,Les auteurs ont constaté qu'UniMoMo surpassait les méthodes traditionnelles existantes en matière d'évaluation complète basée sur CBGBench.
Plus précisément, UniMoMo a obtenu des scores globaux plus élevés en termes de distribution de sous-structure (types atomiques, groupes fonctionnels, etc.), de rationalité des propriétés chimiques (QED, LogP, SA, etc.), de qualité de la structure géométrique (distribution de la longueur/de l'angle de liaison et taux de conflit atomique, etc.) et de score d'interaction (amarrage de Vina) (veuillez vous référer au texte original pour les résultats expérimentaux complets). Il est important de noter qu’UniMoMo(all), qui est formé sur tous les types de molécules, montre des améliorations significatives dans toutes les dimensions d’évaluation par rapport à la version à domaine unique formée uniquement sur des données de petites molécules. Cela montre queMême dans le scénario de petite molécule avec la structure moléculaire la plus flexible et les types les plus divers, le modèle unifié peut toujours transférer les lois géométriques et les modèles d'interaction d'autres types moléculaires, améliorant ainsi la rationalité de la conformation du monomère et la disposition relative de l'espace de poche de la petite molécule.Ce phénomène vérifie une fois de plus le concept de base d'UniMoMo : les contraintes géométriques et les mécanismes de liaison entre différentes molécules ont des modèles partageables, et la modélisation unifiée peut stimuler efficacement ce potentiel.
En combinant les résultats expérimentaux des trois types de tâches, UniMoMo présente des avantages très cohérents : le modèle unifié formé à l'aide de données d'espèces moléculaires croisées surpasse le modèle de génération à domaine unique existant dans ses tâches respectives et présente des capacités évidentes améliorées par rapport à UniMoMo formé uniquement avec des données à domaine unique. Ce phénomène montre que les tâches apparemment différentes de la conception moléculaire ont en réalité un degré élevé de points communs dans les contraintes physiques et chimiques sous-jacentes et dans les lois géométriques spatiales.La stratégie de modélisation unifiée d'UniMoMo capture et amplifie ce point commun, permettant ainsi un transfert intertâches et une amélioration complémentaire.Ces résultats vérifient non seulement l’efficacité d’UniMoMo, mais fournissent également un solide soutien empirique pour la construction d’un système de génération moléculaire unifié plus puissant à l’avenir.
Études de cas GPCR
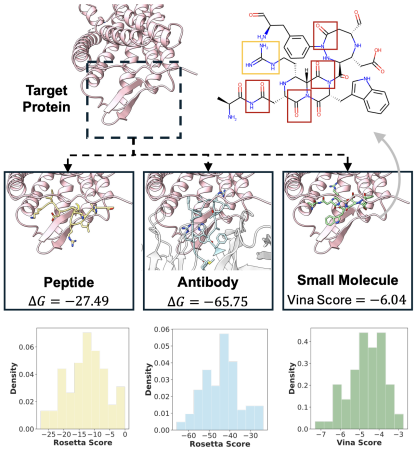
Dans le cadre d'une étude de cas, les auteurs ont sélectionné l'une des cibles médicamenteuses les plus importantes chez l'homme, le récepteur couplé aux protéines G (GPCR), pour évaluer la capacité d'UniMoMo à générer différents types de molécules (peptides, anticorps, petites molécules) sur le même site de liaison. Les peptides, anticorps et petites molécules générés par UniMoMo présentent tous une bonne distribution sous les champs de force couramment utilisés pour l'évaluation de l'énergie de liaison (score Rosetta ΔG, Vina).Ce qui est encore plus surprenant, c'est que la structure de petite molécule générée simule également spontanément des groupes fonctionnels similaires aux chaînes latérales d'acides aminés naturels, qui sont utilisés pour construire des liaisons hydrogène et former des interactions clés avec la cible. De plus, les petites molécules empruntent également des configurations géométriques locales aux peptides et aux anticorps, telles que les connexions amides sur le squelette moléculaire, qui leur permettent de remplir efficacement les poches de liaison qui sont à l'origine plus adaptées aux grosses molécules. Ce cas démontre de manière éclatante la capacité d'UniMoMo à emprunter à travers les modalités et à s'adapter automatiquement aux poches de liaison dans les tâches réelles, et reflète son potentiel à comprendre en profondeur l'interaction entre les cibles et les molécules et les contraintes géométriques internes des molécules au niveau structurel tridimensionnel.
Exploration future
Bien qu'UniMoMo ait démontré de fortes capacités de génération unifiée dans de multiples types et tâches moléculaires, les auteurs ont également souligné qu'il existe encore de nombreuses possibilités futures qui méritent d'être explorées dans cette direction.
Les travaux actuels se concentrent principalement sur la modélisation des acides aminés naturels et des fragments moléculaires courants, qui peuvent être étendus à des formes de médicaments plus complexes telles que les acides aminés non naturels, les peptides/anticorps post-modifiés, les molécules cycliques, etc., couvrant ainsi une gamme plus large d'espaces moléculaires candidats. Le concept de modélisation unifiée offre également la possibilité d’étudier la contrôlabilité et l’interprétabilité du modèle, et devrait favoriser davantage le développement de modèles génératifs vers des plateformes de conception moléculaire plus fiables et plus pratiques. En résumé, l’introduction d’UniMoMo fournit non seulement un cadre génératif général et puissant pour les tâches de conception moléculaire, mais ouvre également une nouvelle direction pleine de potentiel pour la découverte de médicaments pilotée par l’IA.








