Command Palette
Search for a command to run...
AI-driven Quantum Refinement: Carnegie Mellon University and Others Propose AQuaRef, the First to Use Quantum Mechanical Constraints to Refine a protein's whole-atom model.
To understand the molecular mechanisms of life processes, we first need to see the three-dimensional structure of biological macromolecules.Determining atomic-level structures is a core task of structural biology and an important foundation for understanding protein function, revealing genetic regulatory mechanisms, and developing targeted drugs.Whether it's protein-catalyzed reactions, nucleic acid transmission of genetic information, or antibody recognition of antigens, these key biological processes all rely on precise structural models for explanation.
Currently, cryo-electron microscopy and X-ray crystallography are the main experimental techniques for resolving the structures of biological macromolecules, and a large amount of high-resolution structural data has been accumulated. In recent years, computational prediction methods, represented by AlphaFold and RoseTTAFold, have also made significant progress, providing efficient tools for structure modeling. However, experimental analysis still plays an irreplaceable role in discovering unknown structure types and resolving complex interactions.In the experimental structure resolution process, atomic model refinement is a key step that is close to the final stage. Its goal is to construct a molecular model that conforms to the laws of stereochemistry and fits the experimental data as closely as possible.Current mainstream refinement software, such as CCP4 and Phenix, mainly relies on stereochemical constraints in standard databases to maintain reasonable bond lengths and bond angles and reduce interatomic conflicts.
However, such constraint systems still have significant limitations. They primarily target covalent structures and lack a systematic description of important non-covalent interactions such as hydrogen bonds and π-packing. At low resolution, this can lead to models deviating from the true chemical state. When novel ligands or unique connections appear in the structure, manual parameter definition is required for refinement. Furthermore, reasonable geometric deviations caused by local chemical environments may be misinterpreted as anomalies by the constraint system and forcibly corrected. Theoretically,Quantum mechanics can describe intermolecular interactions more accurately, but biological macromolecules typically contain thousands or even tens of thousands of atoms, making full quantum computing extremely expensive.Therefore, most existing studies are limited to local areas such as ligand binding sites.
To address this issue, a joint research team from Carnegie Mellon University, the University of Wrocław in Poland, and the University of Florida, among other universities,An AI-driven quantum refinement method, AQuaRef, is proposed.This method is based on AIMNet2 machine learning of atomic potential functions and has been customized for the refinement task. While approaching the computational efficiency of classical force fields, it can better approximate the results of quantum mechanical calculations, providing a new technical path for the all-atomic quantum refinement of biological macromolecules.
The related research findings, titled "AQuaRef: machine learning accelerated quantum refinement of protein structures," have been published in Nature Communications.
Research highlights:
* AQuaRef, based on the AIMNet2 machine learning potential function, achieves quantum refinement of a full protein atom model for the first time.
* In tests of 61 low-resolution X-ray and cryo-electron microscopy models, AQuaRef outperformed 57 models.
* In the cases of short hydrogen bonds in DJ-1 and YajL proteins, AQuaRef can determine proton positions consistent with experimental evidence without human intervention.

Paper address:https://www.nature.com/articles/s41467-025-64313-1
Follow our official WeChat account and reply "AQuaRef" in the background to get the full PDF.
A dataset of 1 million samples for training the potential function in peptide machine learning.
This study aims to construct a parameterized model of the potential function for a peptide system using machine learning.Therefore, the dataset design needs to systematically cover three dimensions: chemical composition, conformational space, and intermolecular interactions.
From a chemical perspective,Researchers constructed a small peptide database in the form of SMILES strings, covering 20 standard amino acids, 11 protonation states, 3 N-terminal modifications, and 4 C-terminal modifications.Building upon this foundation, all single and dipeptides were enumerated, and a subset of tripeptides and tetrapeptides were randomly selected. Additionally, peptides linked by disulfide bonds and their selenized analogs were generated. To fully cover the conformational space, the researchers used OpenEye Omega software for intensive torsion angle sampling without imposing restrictions on chiral centers, thus enabling the model to be applicable to D-type, L-type, and mixed stereochemical peptide systems.
Complexes consisting of 2-4 peptides were constructed, and their spatial orientation was randomly adjusted to simulate intermolecular interactions. The entire data generation process did not reference natural sequences or experimental structures to avoid potential data leakage. To control computational scale, the total number of atoms (including hydrogen) in all peptides and their complexes was limited to 120.
After obtaining the initial conformationResearchers first used GFN-FF force fields to conduct molecular dynamics simulations to sample non-equilibrium structures.It maintains the overall configuration close to the initial input by constraining it with Cartesian coordinates, while releasing the torsion angle and intermolecular degrees of freedom.
Subsequently, a query-by-committee active learning strategy was introduced: First, 500,000 initial samples were randomly selected to train an ensemble system consisting of four models. Then, four iterations were performed. In each iteration, samples were selected based on the uncertainty of the models' predictions of energy and atomic forces, and these high-uncertainty structures were added to the training set after DFT calculation. The final iteration further introduced uncertainty-guided optimization, prioritizing boundary structures with high prediction uncertainty but low energy. Through this process, a training set of approximately 1 million samples was finally obtained, with an average of approximately 42 atoms.
In addition to theoretically generated data, researchers also screened experimental structures from the RCSB and EMDB databases for model validation. Screening criteria included: single conformation models containing only proteins, 1,000–10,000 non-hydrogen atoms, resolution of 2.5–4 Å, MolProbity conflict score of less than 50, and bond length and bond angle deviations not exceeding 4 times the standard values.
AQuaRef: AI-Driven Quantum Refinement Approaches for Macromolecular Systems
AQuaRef first performs an integrity check on the input atomic model. For missing atoms in the structure, the program attempts to automatically fill them in. However, this process can sometimes introduce new steric hindrance conflicts, especially if the original model does not contain hydrogen atoms. If the missing atoms are critical structures such as main-chain atoms, the model cannot continue with quantum refinement; if significant steric conflicts or severe geometric anomalies are detected, rapid geometric regularization using standard stereochemical constraints is performed first to eliminate the problem with minimal adjustments to the atomic positions.
For crystallographic data, refinement also needs to take into account unit cell symmetry and periodic interactions.Specifically, the program expands the model into a supercell based on the space group symmetry operator and then truncates it, retaining only the symmetric copies whose distance from the main copy atom is within a set range. This process is typically unnecessary in cryo-electron microscopy structures.
After completing atomic supplementation and model expansion, the system enters the standard refinement process of the Q|R software package. The core architecture of AQuaRef is basically the same as the basic AIMNet2 model, but several key adjustments have been made for the structural refinement task.
First, the model does not explicitly compute long-range Coulomb and dispersive interactions, but is directly trained to reproduce the total DFT-D4 energy.This is because, under the CPCM implicit solvent model, the Coulomb interaction is difficult to estimate accurately using only the atomic charge, and the long-range interaction is significantly shielded by the polarizable continuous medium. In addition, the long-range dispersion terms with a cutoff radius of more than 5 Å contribute very little to the key atomic forces in the refinement process, so they can be ignored without affecting the accuracy.
Secondly, the model introduces an explicit short-range exponential repulsion term from GFN1-XTB, which results in better stability when dealing with structures with spatial steric hindrance conflicts.Model training was performed using the energy, atomic force, and Hershfield partial atomic charge calculated by the B97M-D4/def2-QZVP method. Training started with random weight initialization, a batch size of 256, and a total of 1.5 million training steps. All other hyperparameters were retained from the original AIMNet2 settings.
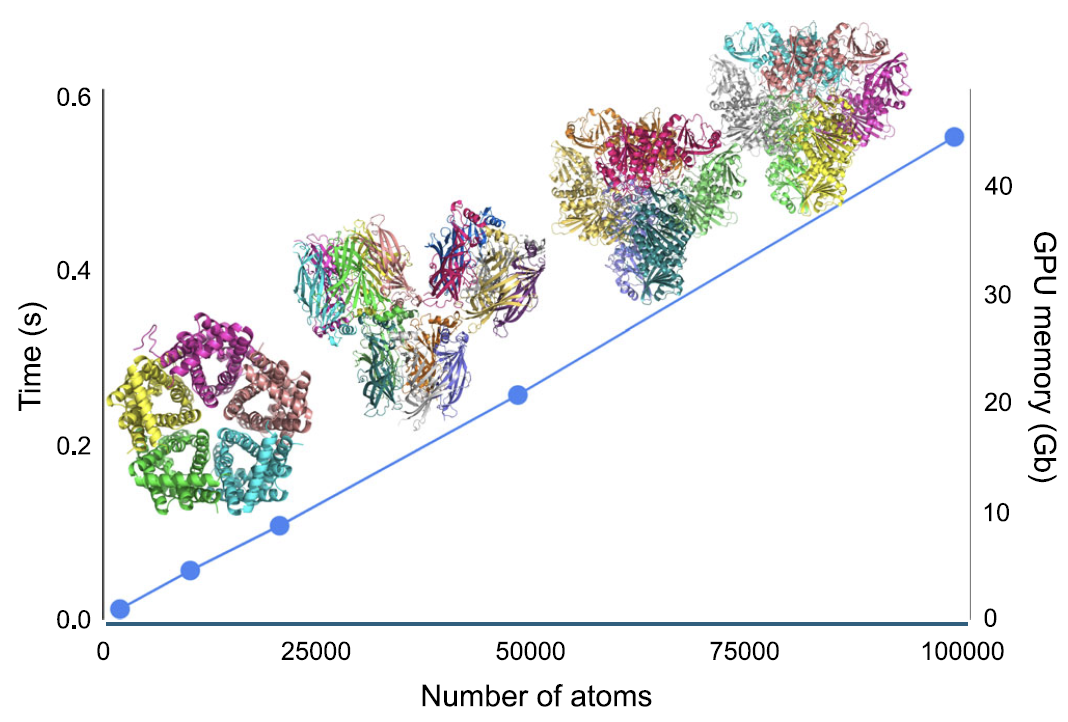
Regarding computational efficiency, as shown in the figure below...In the AIMNet2 framework, the computation time for energy and atomic forces, as well as the peak GPU memory usage, all increase linearly (O(N)) with the number of atoms in the system.For a protein system containing approximately 100,000 atoms, single-point energy and force calculations take only about 0.5 seconds; on a single NVIDIA H100 GPU equipped with 80GB of video memory, models with up to approximately 180,000 atoms can be processed.
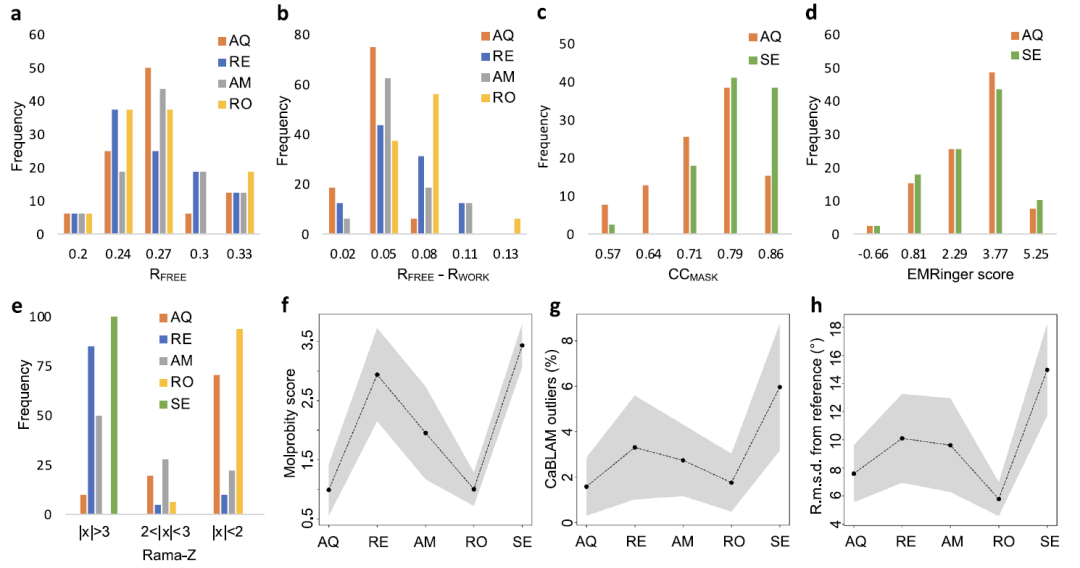
Validated by 41 cryo-electron microscopy and 20 X-ray model analyses, the local structure of AQuaRef was optimized to 2 Å.
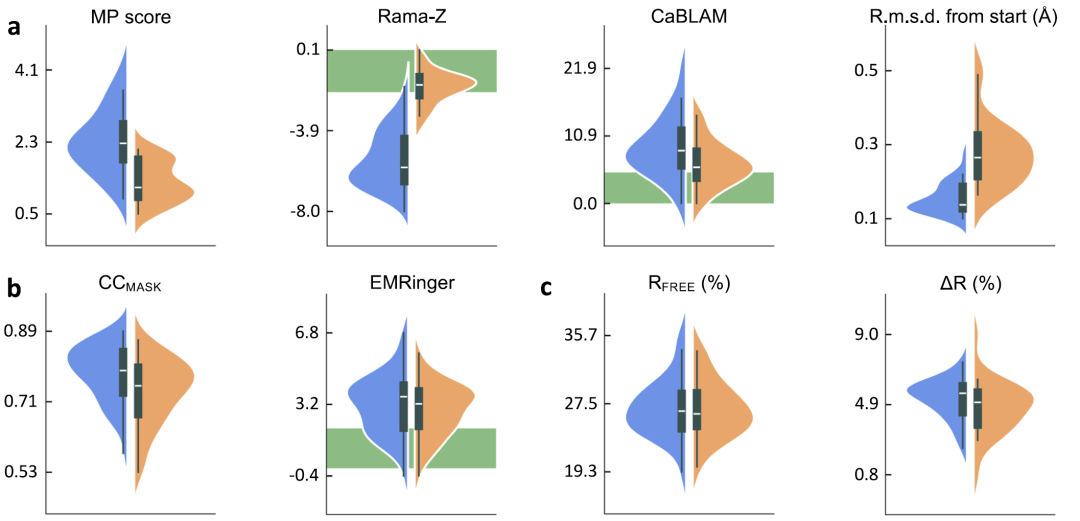
To evaluate the performance of AQuaRef,The researchers constructed a test set that included 41 cryo-electron microscopy models, 20 low-resolution and 10 ultra-high-resolution X-ray models.All 61 low-resolution models are equipped with corresponding high-resolution homologous reference structures. During the refinement process, three types of constraints were set for comparison: AIMNet2 quantum constraints (i.e., AQuaRef), standard geometric constraints, and additional constraints such as hydrogen bonding and secondary structures on top of the standard constraints.
The results are shown in the figure below.The quantum-refined low-resolution model significantly outperforms traditional constraint methods in geometric metrics such as MolProbity score and Ramachandran plot Z-score.Meanwhile, the model's fit to the experimental data remained largely consistent. For X-ray structures, the overfitting was slightly reduced (the difference between Rwork and Rfree was smaller); for cryo-electron microscopy structures, CCmask decreased slightly while the EMRinger score remained essentially unchanged. Combined with the overall improvement in geometric quality, these results suggest that model overfitting may have been reduced.
While adding additional geometric constraints to standard constraints can improve model quality, AQuaRef still yields more reasonable geometries and is closer to the high-resolution reference model. In some cases, the local difference between the standard constraints and the quantum-refined structure can reach 2 Å.
The study also compared AQuaRef with several mainstream refining methods. The results are shown in the figure below. AMBER, Rosetta, and REFMAC5 were selected for X-ray data, while Servalcat was used for cryo-electron microscopy data. Overall,AQuaRef has slightly better Rfree performance and the lowest degree of overfitting.Compared to Servalcat, both have comparable EMRinger scores, but Servalcat has a slightly higher CCmask score.
In terms of geometric quality,AQuaRef performs similarly to Rosetta, but significantly outperforms REFMAC5 and Servalcat.Rosetta has a slightly higher overall fit with the reference model, which may be related to the larger convergence radius resulting from its non-gradient optimization strategy. Furthermore, AQuaRef and Rosetta can both generate reasonable hydrogen bond geometries, followed by AMBER, while REFMAC5 and Servalcat are basically unable to accurately recover these details.
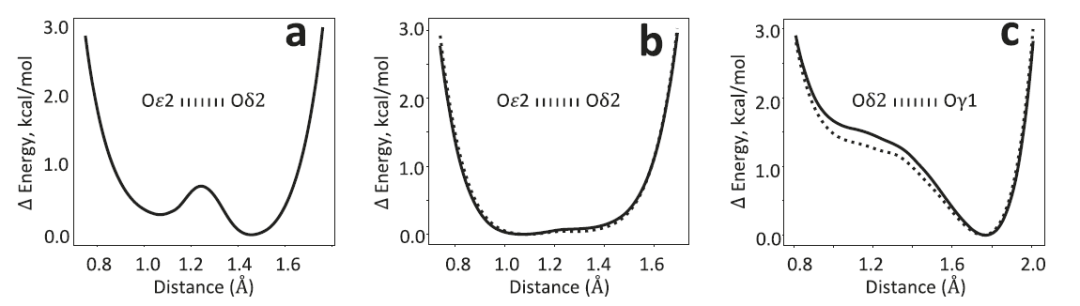
In tests on short hydrogen bond systems, researchers used the Parkinson's disease-associated protein DJ-1 and its homolog YajL as examples to examine AQuaRef's ability to handle protonated states. Traditional refinement methods, constrained by database stereochemistry, often cause bond lengths to deviate from their true values.When using the symmetric diprotonated structure as the initial model for AQuaRef refinement, the resulting proton positions and bond geometry are consistent with the unconstrained refinement results.With the addition of conventional constraints, bond lengths are pulled toward the deprotonated standard values in the database. When experimental data is truncated to 2 Å resolution, significantly reducing atomic details, AQuaRef can still recover a structure almost identical to the original 1.15 Å data, while conventional constraint refinement deviates further from the true configuration. AQuaRef positions the proton at the Oδ2 atom of the D24 residue in DJ-1, a result supported by both energy calculations and differential electron density maps.
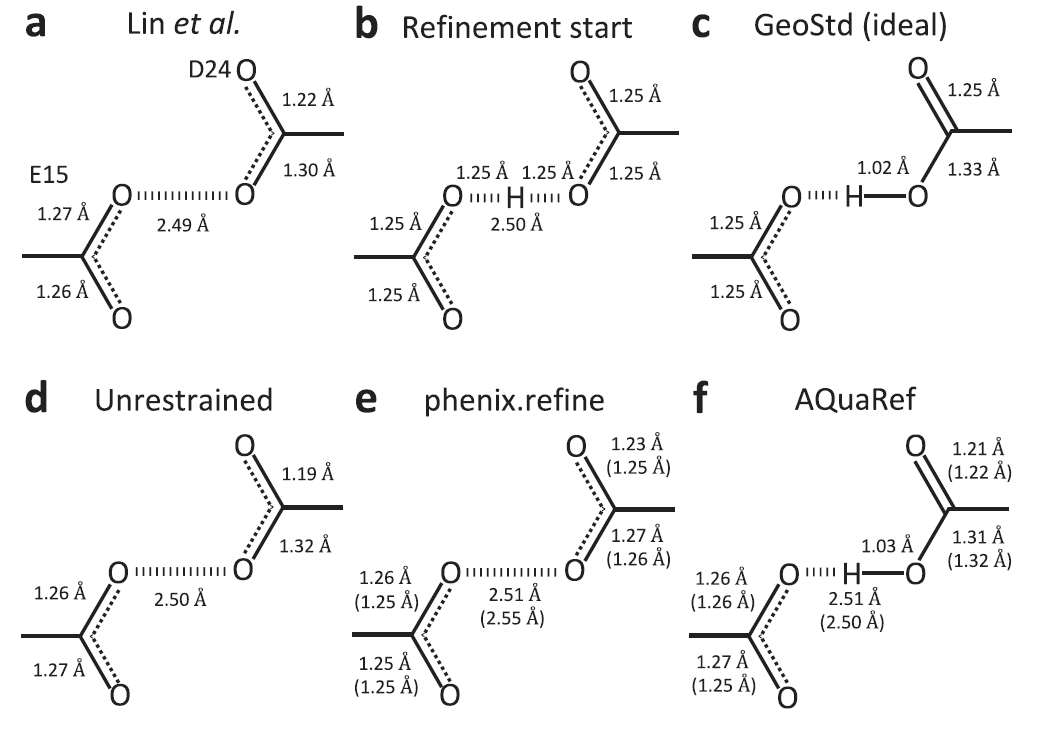
In the YajL protein, the AQuaRef refinement results for the two E14/D23 short hydrogen bonds are consistent with the unconstrained refinement, indicating that the proton is shared by both D23 and E14, exhibiting typical low-barrier hydrogen bond characteristics. This differs from the case in DJ-1, where the proton is mainly located on a single oxygen atom. The energy distribution given by AIMNet2 shows a relatively flat potential energy surface, meaning that the proton position can be freely adjusted under the constraints of experimental data. Simultaneously, the differential electron density plot shows significantly higher peaks than 3σ near the hydrogen atom, providing further evidence for this structural interpretation.
Breakthroughs in Industry-Academia-Research Collaboration in the Field of Protein Quantum Refinement
In the cutting-edge fields of protein quantum refinement, machine learning potential function construction, and atomic model optimization, multiple research teams have been continuously exploring this direction and have made a series of advances. For example,The neural network method nn-tm fcc developed by the Oxford University team can construct high-precision potential energy surface models of residue fragments with near-full quantum mechanical accuracy.The root mean square errors of energy and atomic force calculations are controlled within 1.0 kcal/mol and 1.3 kcal/(mol·Å), respectively. Using this method, energy and atomic force calculations for 15 representative proteins can be completed in just 10 to 100 seconds, which is thousands of times faster than traditional quantum mechanics calculations.
Paper title: Improved protein structure prediction using potentials from deep learning
Paper link:https://www.nature.com/articles/s41586-019-1923-7
Another German collaborative team proposed the BF-DCQO quantum algorithm, which combines a non-variable iterative strategy with the IonQ ion trap quantum computing system.The computation time for a 3D folding problem involving 12 amino acids has been reduced from 72 hours with a traditional GPU cluster to approximately 4.3 minutes.The speed increase also reached a thousandfold.
Thesis title: Bias-field digitized counterdiabatic quantum algorithm for higher-order binary optimization
Paper link:https://www.nature.com/articles/s42005-025-02270-3
Overall, the combination of quantum mechanical methods, machine learning potential functions, and experimental structural data is providing a new technical approach for refining the structures of biological macromolecules, and is expected to play a more stable role in scenarios such as low-resolution structural modeling, ligand binding mode analysis, and functional site research.