Command Palette
Search for a command to run...
From Quartz to Ferroelectric Materials, Harvard University Proposes an Equivariant Machine Learning Framework to Accelerate large-scale Electric Field Simulation of Materials

As a cutting-edge research direction in the field of modern materials science, computational materials science undertakes the key mission of analyzing the microstructure of materials and predicting their macroscopic properties. Based on the first principles and basic physical laws such as quantum mechanics, this discipline is committed to accurately predicting the experimentally measurable properties of real materials, thereby gaining a deep understanding of the response mechanism of materials under external stimuli. These response characteristics include linear, nonlinear and coupling effects.It is the core element that determines the functional performance of dielectrics, ferroelectrics, multiferroic materials and piezoelectric materials.
Currently, the first-principles electronic structure method based on density functional theory (DFT) is an important means to study the properties of materials.However, due to the fact that the computational cost increases exponentially with the size of the system, it can only handle small-scale systems.This has greatly restricted the systematic research on complex material systems. In recent years, the introduction of machine learning methods has brought breakthroughs in this field. By building data-driven models, it has shown great potential in predicting material polarization, Born charge, polarizability, dielectric constant, spectral characteristics, etc., and has been successfully applied to molecules, liquid water and solid material systems.
However, most existing machine learning methods still have limitations. For example, it is difficult for independently designed models to ensure the strict implementation of physical symmetries and conservation laws. Some single-model solutions that attempt to integrate dielectric properties, energy, and force calculations face challenges when extended to actual systems with periodic boundary conditions and polarization multi-values. Although some studies have constructed potential energy surfaces by training atomic forces under different electric fields and indirectly deduced polarization characteristics to circumvent the difficulty of training multi-valued polarization data,However, this method relies on implicit derivative calculations, which may reduce prediction accuracy, and the high cost problem of large-scale DFT data acquisition has not been fundamentally solved.
To address the above challenges, Harvard University and Robert Bosch LLC, a subsidiary of the German Bosch Group in the United States, jointly developed a unified differentiable learning framework for electrical responses.This framework can simultaneously learn the generalized potential energy and its response function to external stimuli in a single machine learning model.By defining the response function as the derivative of the generalized potential energy with respect to the atomic coordinates and perturbation parameters, and utilizing the precise mathematical relationship between the two, and strictly satisfying physical constraints such as momentum conservation and the Born charge acoustic summation rule, the inherent defects of traditional independent models are overcome, opening up a new path for high-precision research on the dielectric and ferroelectric properties of crystalline, disordered and liquid materials.
The relevant research results have been published in the internationally renowned journal Nature Communications under the title "Unified differentiable learning of electric response".
Research highlights:
* The first unified machine learning framework to achieve multi-dimensional conservation guarantees for momentum, electric enthalpy, etc. based on the precise differential relationship between generalized potential energy and observable response quantities.
* Develop an equivariant neural network model to break through the bottleneck of ferroelectric hysteresis simulation at the million-atom level and accurately analyze the domain nucleation and one-dimensional expansion dynamics of polarization switching.
* Solve the problem of multi-valued polarization training, and combine first principles to achieve cross-scale high-precision prediction of dielectric and ferroelectric properties in the α−SiO₂/BaTiO₃ system.

Paper address:
https://go.hyper.ai/18TWg
More AI frontier papers:
α−SiO₂ and BaTiO₃ data experiments, training set construction and key parameter derivation
This study conducted data experiments on two materials: α-SiO₂ and BaTiO₃.The model performance is verified by constructing training sets and validation sets.
In the training data generation phase, 200 frames of atomic configurations of α−SiO₂ were extracted from NVT classical MD simulations at 300 K and 600 K (Vashishta potential driven, each running for 100 ps, 1 ps interval sampling). For BaTiO₃, 75 frames (300 K–400 K, 60 frames of original structure and 15 frames of domain wall structure) were collected through active learning dynamics of FLARE code.
For each set of data, the energy, force, and polarization in the absence of an electric field were calculated by DFT, and the Born charge and polarizability were derived using the finite difference method under a small electric field of 0.36 MV/cm to ensure the linear response range of polarization and electric field.
Material response prediction framework based on equivariant neural network
In this study, researchers successfully developed an innovative machine learning framework that aims to achieve unified learning of generalized potential energy and response functions. This framework strictly follows the mathematical principles of Taylor expansion.Ingeniously, the learning tasks of generalized potential energy and its derivatives are carried out simultaneously within a unified model framework.This allows for accurate prediction of the material's response characteristics.
The core concept of this model is thatBy taking the derivative of the generalized potential energy with respect to its key variables (such as atomic positions and external fields), the corresponding response characteristics can be automatically generated for each atomic configuration.It covers many aspects such as force, polarization and Born charge. This design not only ensures that physical symmetry and conservation laws such as conservation of momentum and conservation of electric enthalpy are accurately implemented, but also significantly improves the prediction accuracy and reliability of the model.
In the training phase of the model,The researchers did this by adding parameters describing the system's perturbations to the input, allowing the model to differentiate these parameters.In this way, training can be performed on additional physical quantities. At the same time, the researchers also minimized a comprehensive loss function that fully integrates the contribution factors of various response characteristics such as energy, force, polarization, and Born charge. This training mode is closely related to the concept of Sobolev training. Specifically, each term that makes up the loss function is composed of the difference between the gradient corresponding to the training label and the energy.
In order to accurately capture the nonlinear dependence of the response characteristics on multiple fields and the coupling characteristics,The researchers used a neural network architecture and a powerful technique called gradient backpropagation.Deeply learn the intricate network of relationships between generalized potentials and their inputs.
In the process of building the model architecture,Researchers rely on the Allegro platform.Taking full advantage of the high precision and outstanding data efficiency of equivariant neural networks,At the same time, by strictly following the local design concept, the model has achieved excellent scalability. In terms of the model input design, as shown in the figure, the researchers cleverly merged the electric field E with the atomic position r_i, which enables the model to simultaneously carry out the learning tasks of electric enthalpy U, force F_i, polarization P, Born charge Z_i and polarizability α.
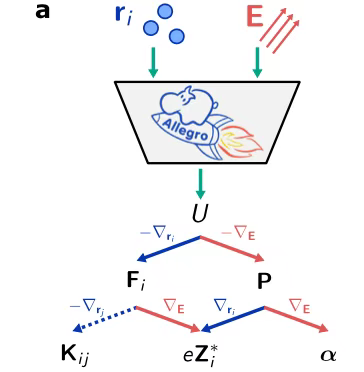
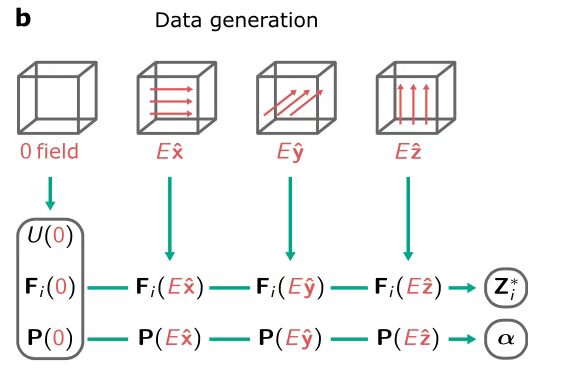
In the process of generating training data,As shown in the figure below, the researchers used DFT calculations to obtain data in the area around zero electric field, and used the finite difference approximation method to determine the values of Born charge and polarizability. After training, the model has the ability to output enthalpy, and then derive a series of key parameters such as force, polarization, Born charge and polarizability by calculating the first and second order derivatives of the output enthalpy.
It is worth mentioning that the researchers have specially developed a corresponding interface so that the model can be seamlessly integrated with the LAMMPS software, thereby strongly supporting large-scale structural relaxation and machine learning molecular dynamics (MLMD) simulations under electric field conditions.
Multi-scenario verification of model accuracy
To verify the performance of the model, the research conducted experiments in two major directions: material vibration and dielectric properties, ferroelectric hysteresis, and dipole dynamics.Taking α−SiO₂ and BaTiO₃ as research objects, the accuracy and applicability of the model in different scenarios are deeply explored.
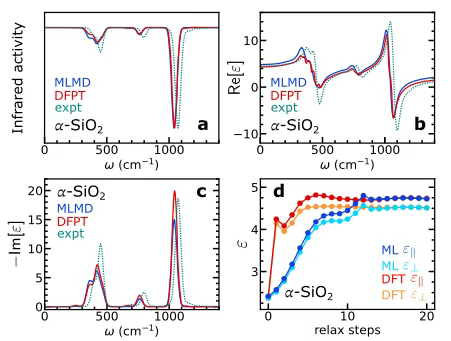
In the study of vibration and dielectric properties,The researchers chose α−SiO₂ as a typical research object, extracted 200 frames of data of 72 atoms from the molecular dynamics simulation of classical potential energy, and trained the corresponding machine learning model based on this. In order to further explore the accuracy of the model, a supercell containing 24,696 atoms was constructed. After 10 picoseconds of equilibrium under the NVE ensemble, a 200 picoseconds machine learning molecular dynamics (MLMD) simulation without electric field was carried out. The infrared spectrum was calculated by polarization dynamics, and the polarization and polarizability dynamics were analyzed to determine the frequency-dependent dielectric constant. Finally, the results were compared with the calculation results based on density functional perturbation theory (DFPT) and experimental data.
As shown in Figure ac below,The results of MLMD and DFPT were highly consistent.In addition, when studying the shielding effect of electrons and ions in the presence of an electric field, the researchers performed structural relaxation under a finite electric field based on the original bulk structure of α-SiO₂. The static dielectric constant was determined by calculating the polarization difference between the electric field and the absence of an electric field. The results are shown in Figure d below. The high-frequency and static dielectric constants obtained by the model are basically consistent with the DFT values.This fully demonstrates that the model can accurately capture the contribution of the electric field to the electronic structure, verifying its effectiveness in finite electric field dynamics simulations.
At the same time, in the training verification of Born charges, it was found that if the model is not trained on Born charges, the accuracy of vibration and dielectric response at low frequencies will be significantly affected, especially when data is limited, and the computational cost of simulation under electric fields will increase.This finding highlights that training on Born charges is a key advantage of the model.
In the study of ferroelectric hysteresis and dipole dynamics,The researchers focused on BaTiO₃ perovskite and used 75 frames of data extracted by active learning dynamics, with 135 atoms per frame, to train the machine learning model. The ferroelectric hysteresis of the 135-atom supercell was calculated at zero temperature, and the results are shown in Figure a below.The ferroelectric hysteresis obtained by MLMD simulation is highly consistent with that obtained by DFT calculation, which strongly verifies the reliability of the model.
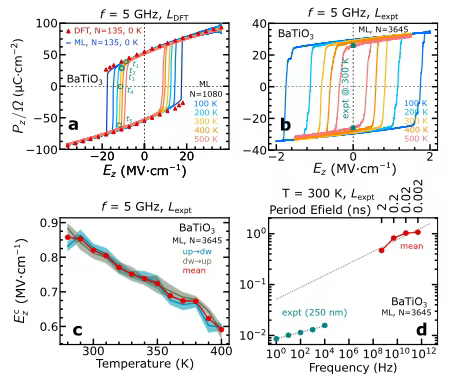
To further evaluate the effect of temperature on the ferroelectric response, the researchers conducted MLMD in the NVT ensemble under a 5 GHz sinusoidal electric field. The study found that, as shown in Figures bc above,As the temperature increases, the intrinsic coercive field decreases, while the spontaneous polarization is relatively less affected by temperature.Whether at zero temperature or finite temperature, the hysteresis curves show electric field sign symmetry, which is consistent with the characteristics of polarization conservative vector fields. In addition, when using a 3,645-atom supercell with experimental lattice parameters for research, the spontaneous polarization is consistent with the experimental results, and the coercive field is also closer to the experimental value, while verifying the model's ability to extrapolate under supercells with different atomic numbers and lattice parameters.
When investigating the effect of electric field frequency on ferroelectric hysteresis,MLMD of a 3,645-atom supercell at 300 K shows that the coercive field decreases with decreasing frequency. Although the extrapolated value is still about an order of magnitude different from the experimental value, this complex computational task highlights the powerful computational capabilities of the model and its excellent scalability to million-atom systems.
Material model research: collaborative progress between academia and industry
In the field of materials science, both academia and the business community have made great efforts to promote the continued development of research related to material models.
For the academic community, many universities and research teams have achieved remarkable results. Researchers at the University of Rochester in the United States have developed a machine learning model that can analyze the massive data generated by X-ray diffraction (XRD) experiments to accelerate material innovation. The model is trained with experimental data of inorganic materials under different experimental conditions and crystal properties, and is classified and optimized according to the Bragg law. The Chemeleon model proposed by the Imperial College of London uses generative AI to navigate based on the material structure characteristic data set, learn from text descriptions and three-dimensional structure data, and achieve sampling of chemical composition and crystal structure. The collaborative team of Seoul National University in South Korea and Fordham University in the United States used a large language model (LLM) to predict the synthesizability of new materials and explained the basis for the prediction, showing good performance and interpretability. The team of Professors Wang Jinlan and Ma Liang from the School of Physics at Southeast University in China and the team of Professor Wang Xinran from Nanjing University proposed new ideas in the research of two-dimensional materials, realizing the number-controlled epitaxial growth of centimeter-level uniform double-layer MoS₂ thin films, and proposed methods to improve the performance of contact interfaces through computational simulation, successfully achieving ultra-low contact resistance.
The business community is not to be outdone and is actively engaged in the innovation and application of material model technology. Apple uses machine learning models to optimize metal alloy formulas for product shell manufacturing, improve material strength and durability, and reduce costs. BASF develops advanced material models to simulate material properties, accelerate the development of new plastics and coatings, and improve product competitiveness. The MatterGen model launched by Microsoft generates material structures that meet design requirements through a unique diffusion model architecture, which has significant advantages over traditional methods. It has also cooperated with the SIAT team of the Chinese Academy of Sciences to obtain a new material TaCr₂O₆.
These cutting-edge explorations and innovative practices are intertwined, continuously pushing material model research forward. In the future, as research continues to deepen and technology iterates and upgrades, material models will achieve breakthroughs and applications in a wider range of fields, laying a solid foundation for scientific and technological development.
Reference articles:
1.https://mp.weixin.qq.com/s/mctu0DOWO_OieLnOgp93Rw
2.https://mp.weixin.qq.com/s/I-UZTyUFSWwXlf1LCmwjRQ
3.https://mp.weixin.qq.com/s/Ox62ut3IJcUWsLC7sF100Q
4.https://mp.weixin.qq.com/s/VlPb8zSghVVxnPNl-WzqBA
5.https://plastics-rubber.basf.com/global/en/performance_polymers/services/service_ultrasim/Material-Modeling








