Command Palette
Search for a command to run...
홍콩중문대학교, 저장대학교, 마카오공업대학교 연구팀은 생물학적 의미론과 화학적 정밀도를 통합하여 350%를 이용한 용혈성 질환 예측 정확도를 향상시키는 일반적인 프레임워크인 Bi-TEAM을 제안했습니다.

생화학 및 분자 공학 분야에서 특성 학습은 분자 기능 규명 및 치료 분자 발굴을 위한 핵심 기술로 점차 자리 잡고 있습니다. 내장된 특징의 품질은 펩타이드 특성 예측 및 신규 설계와 같은 후속 작업의 성능 한계를 결정하는 중요한 요소입니다. 생물학적 기능과 화학적 특성을 연결하는 핵심 분자인 펩타이드의 구조 및 기능 모델링은 신약 개발에 있어 매우 중요한 가치를 지닙니다.최근 몇 년 동안 비고전적인 아미노산의 도입으로 펩타이드의 기능적 범위가 크게 확장되고 안정성과 생체 이용률이 향상되었지만, 복잡한 화학적 변형은 기존 모델링 방법에 새로운 과제를 가져왔습니다.생물학적 진화 정보와 화학적 합리성을 동시에 모델에 통합하는 방법은 이 분야에서 시급히 해결해야 할 핵심 과제로 떠오르고 있다.
현재 펩타이드 모델링은 주로 두 가지 기술적 경로를 통해 수행됩니다.한편으로는,ESM 및 ProtT5와 같은 단백질 언어 모델은 대규모 시퀀스 사전 학습을 통해 생물학적 맥락과 진화 정보를 포착하여 후속 작업에 활용 가능한 생물학적 표현을 제공합니다.반면에,비고전적 아미노산 변형 문제를 해결하기 위해 연구진은 원자 수준의 단어 분할을 통해 화학적 세부 사항을 포착하는 화학 언어 모델을 사용하여 화학적 수준에서 단백질 모델의 한계를 보완했습니다.
하지만 두 유형의 모델 모두 고유한 한계를 가지고 있습니다. 단백질 언어 모델은 자연 아미노산 문자 집합에 의해 제한되어 비고전적인 잔기를 처리하기 어렵습니다. 어휘를 근사화하거나 확장하는 기존 방법은 종종 편향을 유발하거나 의미적 희소성을 초래합니다. 반면 화학 언어 모델은 전반적인 생물학적 맥락을 무시하고, 밀집된 단어 분할은 맥락 범위를 쉽게 벗어나 긴 서열 모델링에 적용하기 어렵습니다. 일반적인 모델 또한 도메인 편향의 문제점을 가지고 있습니다.
앞서 언급한 문제들을 해결하기 위해 홍콩중문대학교는 마카오공업대학교, 저장대학교, 중남대학교 부속 샹야제2병원, 중국전자과학기술대학교와 협력하여 선택적 융합 모델링 패러다임을 제안했습니다."화학적 변이는 생물학적 의미 공간의 국소적 교란"이라는 이해를 바탕으로, 국소적인 화학적 변이를 전체 단백질 배경에 주입하기 위한 일반적인 프레임워크인 Bi-TEAM이 설계되었습니다.
이 프레임워크는 생물학적 표현을 의미론적 기반으로 활용하고, 화학적 신호의 적응적 주입을 통해 생물학적 진화 정보와 화학적 논리를 효과적으로 융합합니다. Bi-TEAM은 다양한 작업에서 최첨단 기준 모델들을 일관되게 능가합니다. 골격 유사성을 기반으로 한 엄격한 데이터 분할 조건에서 매튜스 상관 계수는 최대 661 TP3T 향상되었고, 용혈 예측 작업에서는 정확도가 3501 TP3T 향상되었습니다.
"Bi-TEAM: 화학적으로 변형된 생체 분자를 위한 통합된 교차 규모 표현 학습 프레임워크"라는 제목의 관련 연구 결과는 arXiv에 사전 공개 논문으로 게시되었습니다.
연구 하이라이트:
Bi-TEAM 프레임워크는 다중 스케일의 생화학적 특성을 적응적으로 통합하고 효율적인 펩타이드 설계를 위한 고정밀 사전 모델 역할을 할 수 있습니다.
* 연구진은 3개 생화학 분야에 걸쳐 10개의 다양한 데이터 세트를 사용하여 Bi-TEAM을 종합적으로 평가했으며, 7가지 주요 예측 작업에서 최첨단(SOTA) 성능을 달성했습니다.
* 본 모델은 예측 및 생성 작업에서 두 가지 획기적인 성과를 달성하여, 엄격한 골격 유사성 분할 조건에서 MCC를 66%만큼 향상시키는 동시에 세포 투과성 고리형 펩타이드 설계 성공률을 거의 4배 가까이 높였습니다.

서류 주소:
https://arxiv.org/abs/2603.01873
저희 공식 위챗 계정을 팔로우하고 백그라운드에서 "Bi-TEAM"이라고 답장하시면 전체 PDF 파일을 받으실 수 있습니다.
본 연구에서는 세 가지 주요 생화학 분야와 10개의 다양한 데이터셋을 포괄하는 종합적인 평가를 수행하였다.
본 연구는 세 가지 주요 연구 분야인 변형 펩타이드, 단백질 번역 후 변형(PTM), 천연 단백질을 포괄하는 총 10개의 데이터 세트를 사용하여 속성 예측 및 유도 생성이라는 두 가지 차원에서 속성을 평가합니다.
변형 펩타이드 분야에서는 막 투과성을 예측하는 모델의 능력을 평가하는 데 연구가 집중되고 있습니다.핵심 학습 데이터는 12~46개의 고리 원자를 포함하고 서열 길이 분포가 대략 정규 분포를 따르지만 양쪽 끝에 긴 꼬리가 두드러지는 ProPAMPA 데이터베이스에서 가져왔습니다. 또한, 다수의 천연 및 비고전적 아미노산 잔기를 포함하고 있어 높은 화학적 다양성을 보여줍니다. RDKit을 사용하여 중복 제거를 거친 후,이 서열에는 총 6,876개의 비공액 고리형 펩타이드 서열이 포함되어 있습니다.
모델의 일반화 능력을 평가하기 위해 본 연구에서는 ProCacoPAMPA, CycPeptMPDB v1.2 및 Rezai 데이터셋 등 세 가지 외부 습식 실험 데이터셋을 추가로 도입했습니다. 이 데이터셋들은 다양한 길이와 구조 유형을 가진 고리형 펩타이드 샘플을 포함합니다. 구체적으로는 다음과 같습니다.
프로카코팜파:기존 연구에서 길이가 6과 10인 모든 막관통 고리형 펩타이드 서열을 수집하여 표준화된 데이터 세트로 구축했습니다.
CycPeptMPDB v1.2:56편의 논문을 바탕으로 구축된, 공개적으로 이용 가능한 최대 규모의 비고전적 순환 펩타이드 막 투과성 데이터베이스의 최신 버전에는 8,466개의 레코드가 포함되어 있습니다. 본 연구에서는 ProPAMPA 데이터 세트와 중복되는 샘플을 제거하여 최종적으로 1,230개의 데이터 포인트로 구성된 정제된 하위 집합을 얻었습니다.
레자이:여기에는 11개의 고리형 펩타이드에 대한 수동 막 투과성 데이터가 포함됩니다. 이 데이터는 PAMPA 실험을 통해 얻었으며, 소량 샘플 조건에서 외부 모델 검증에 자주 사용됩니다.
모델의 약물 유사성 및 질병 연관성을 더욱 검증하기 위해,연구진은 PTM 데이터셋을 사용하여 약물 유사성 예측 작업을 수행했습니다.본 연구에 사용된 데이터는 두 가지 범주로 나뉘었습니다. 하나는 약물 등급 데이터 세트이고, 다른 하나는 질병 관련 데이터 세트입니다. 약물 등급 데이터 세트는 주로 긴 단백질 서열로 구성되었으며, 변형 부위는 뚜렷한 장꼬리 분포를 보였습니다. 질병 관련 데이터 세트는 주로 dbPTM 및 GWAS(게놈 전체 연관 연구)와 같은 데이터베이스에서 얻었으며, 변형 부위 분포는 약물 등급 데이터 세트와 유사했지만 서열 길이 범위가 더 넓어 보다 다양한 구조적 맥락을 제공했습니다.
천연 단백질 분야에서 연구자들은 펩타이드 용혈 및 단백질 용해도 변화의 핵심 메커니즘을 규명하기 위해 용해도 및 용혈 예측 작업에서 모델 성능 평가에 집중했습니다. 사용된 데이터 세트는 주로 용혈, 항오염, 용해도 세 가지 범주를 포함합니다. 그중에는 다음과 같은 데이터 세트가 있습니다.
용혈 데이터는 DBAASP v3 데이터베이스에서 가져온 것입니다.이 서열은 전형적인 L형 아미노산으로 구성된 총 9,316개의 염기서열을 포함하고 있습니다.
오염 방지 데이터 세트는 주로 짧은 펩타이드 서열로 구성됩니다.길이는 5~10개 아미노산 잔기 사이에 집중되어 있으며, LogP 분포는 대략 정규 분포를 따른다. 샘플들은 특징 공간에서 양호한 클러스터링 구조를 보인다.
용해도 데이터 세트는 PROSO II로 주석이 달린 단백질 서열에서 추출되었습니다.이 명칭은 단백질 구조 이니셔티브에 대한 회고적 분석을 기반으로 합니다.
Bi-TEAM: 화학적으로 변형된 생체 분자를 위한 통합된 다중 스케일 특성화 학습 프레임워크
Bi-TEAM은 기존 단일 모달 모델의 한계, 즉 전역적인 진화 생물학적 정보와 국소적인 화학 구조적 세부 정보(미세한 화학 공간)를 동시에 포착하는 문제를 해결하는 것을 목표로 합니다. 아래 그림에서 볼 수 있듯이,이 연구의 핵심 아이디어는 진화 생물학적 공간과 화학 구조 공간을 심층적으로 통합하는 이중 관점 표현 시스템을 구축하는 것입니다.이를 통해 비고전적인 아미노산을 포함하는 펩타이드 서열에 대해 더욱 정확한 모델링 기능을 제공합니다.
전반적인 건축적 측면에서 볼 때,이 모델은 단백질 언어 모델이 구축한 생물학적 공간을 의미론적 기반으로 사용하며, 대규모 자연 시퀀스에서 학습된 진화 규칙과 문맥적 관계를 최대한 활용합니다.한편, 원자 수준에서 구조 정보를 포착하기 위해 화학 언어 모델(CLM)이 도입되어 화학적 변형을 처리하는 데 있어 단백질 언어 모델(PLM)의 내재적 한계를 보완합니다. 두 모델은 표현 수준에서 서로를 보완하여 입력 시퀀스의 표현력을 공동으로 확장합니다.

Bi-TEAM은 변형된 펩타이드 서열을 처리할 때 두 가지 상보적인 정보 흐름을 사용하여 이를 인코딩합니다.하나는 생물학적 순서 흐름입니다.변형된 아미노산을 구조적으로 가장 유사한 천연 아미노산에 매핑함으로써 단어 분할 테이블 확장을 방지하고 모델링에 사용할 수 있는 진화적 의미론을 보존할 수 있습니다.다른 하나는 셀카와 유사한 표현 방식입니다.이는 변형된 잔류물의 작용기 변화 및 화학 결합 구조를 원자 수준에서 정확하게 기술하는 데 사용되며, 화학 언어 모델에 안정적인 구조 정보를 제공합니다.
듀얼 스트림 인코딩이 완료된 후,이 모델은 위치 인식 단서에 의해 유도되는 이중 게이트 잔류 메커니즘을 사용하여 융합됩니다.생물학적 표현을 의미론적 기반으로 사용하여, 핵심 화학 신호는 게이팅 유닛을 통해 필터링되고 주입되는 반면, 생물학적 특징의 잔여 연결은 보존됩니다. 이를 통해 모델은 훈련 안정성을 유지하면서 전역적인 서열 제약 조건과 국소적인 화학적 변화 사이의 효과적인 상관관계를 설정할 수 있습니다.
응용 분야 측면에서 Bi-TEAM은 뛰어난 다용성을 자랑합니다.수정되지 않은 천연 단백질 서열을 처리할 때, 이 모델은 매핑 및 위치 파악 단계를 직접 생략할 수 있으며, 전체적인 구조를 조정하지 않고도 일반적인 단백질 작업에 적용할 수 있습니다.
훈련 전략 측면에서 보면,본 연구는 "사전 훈련-미세 조정"의 2단계 프레임워크를 사용합니다.먼저, 두 가지 유형의 기본 인코더를 사용하여 자연 단백질 서열과 소분자 화학 물질 코퍼스에 대해 각각 도메인 적응형 사전 학습을 수행했습니다. 그런 다음, 다중 작업 공동 미세 조정을 통해 모델은 다양한 작업 시나리오에서 생물학적 및 화학적 특징의 융합 규칙을 학습하여 전반적인 일반화 능력을 더욱 향상시켰습니다.
Bi-TEAM은 침투성 고리형 펩타이드 설계에서 획기적인 발전을 이루어 성공률을 4.6배 향상시켰습니다.
본 연구는 Bi-TEAM의 미지의 화학적 공간에서의 적용 가능성을 검증하기 위해, 비침습적 약물 전달을 시나리오로 삼아 신생혈관성 연령 관련 황반변성(nAMD)의 세포 투과 치료를 위한 비정형 고리형 펩타이드 설계에 초점을 맞추었습니다. 본 연구는 "예측 기반 분석"의 전 과정을 체계적으로 실험하여, 특성 기반 분자 설계에서 모델의 성능을 평가했습니다.
신생혈관성 황반변성(nAMD)은 노인에서 비가역적 실명의 주요 원인이며, 핵심 병리는 VEGF에 의해 유발되는 맥락막 신생혈관 형성 및 누출입니다. 현재 임상 치료는 주로 아플리버셉트(115kD)와 같은 대분자 항 VEGF 약물의 유리체강내 주사에 의존합니다.하지만 이러한 약물들은 눈의 생리적 장벽을 통과하기 어렵고, 장기간 주사는 합병증 및 환자 순응도 문제를 야기할 수 있습니다.아플리버셉트에 특이적으로 결합하여 장벽 투과를 촉진하는 펩타이드 결합제를 설계하는 것은 비침습적 점안 치료에 새로운 가능성을 열어줄 수 있습니다. 쉽게 분해되고 반감기가 짧은 선형 펩타이드와 달리, 보다 안정적인 구조와 높은 투과성을 지닌 고리형 펩타이드는 이상적인 약물 전달체로 여겨지며, 이것이 연구자들이 고리형 펩타이드 설계에 집중하는 주요 동기입니다.
본 연구는 세포 투과 펩타이드(CPP)의 예측 및 평가를 최초로 수행하여 후속 연구를 위한 토대를 마련했습니다.데이터 세트는 CPPsite2.0, C2Pred, CellPPD 등의 데이터베이스를 통합하여 pLM4CPP 표준 체계에 따라 구축되었습니다. 선별 및 중복 제거 후, 1,399개의 양성 샘플(실험적으로 검증된 침투 펩타이드)과 4,080개의 음성 샘플을 얻었습니다. 비교 모델로는 SeqVec, ESM2, ProtT5와 같은 주요 단백질 임베딩 모델을 사용했으며, 평가 지표로는 ACC, BACC, Sn, Sp, MCC, AUC를 사용했습니다.
결과에 따르면 Bi-TEAM이 모든 지표에서 최고의 성능을 달성했습니다.ACC는 SeqVec 대비 5.521 TP3T, BACC는 ESM2-480 대비 5.881 TP3T, Sn은 12.581 TP3T, Sp는 ProtT5-XL BFD 대비 1.451 TP3T, MCC는 SeqVec 대비 14.681 TP3T, AUC는 ESM2-480 대비 8.451 TP3T 향상되었습니다. 민감도와 MCC의显著한 향상은 이 모델이 실제 침투 펩타이드를 식별하는 데 있어 분명한 이점을 가지고 있음을 나타냅니다.
이를 바탕으로, 특성 기반 순환 펩타이드 생성 실험에 대한 추가 연구가 진행될 것입니다.BoltzDesign1을 기본 프레임워크로 사용하여 두 가지 조건에서 길이 10~20의 고리형 펩타이드 1,000개를 생성했습니다. 첫 번째 조건은 기본 구조적 제약 조건만 사용한 것이고, 두 번째 조건은 생성 과정에서 Bi-TEAM을 추가적인 기울기 가이드로 도입한 것입니다.
성공 기준은 Bi-TEAM 예측 로그 오즈가 0.5보다 큰 것이었습니다. 결과는 다음과 같았습니다...기존 방식으로는 세포 투과성 고리형 펩타이드를 생성하는 데 성공률이 6.71 TP3T에 불과했지만, Bi-TEAM 기반 방식은 이를 30.71 TP3T로 향상시켰습니다.한편, 구조적 품질은 저하되지 않았습니다. 생성된 펩타이드-아플리버셉트 복합체의 평균 pLDDT는 0.82를 초과하여, 모델이 침투력을 향상시키면서도 우수한 구조적 신뢰도와 결합 계면 안정성을 유지했음을 나타냅니다.

유도 메커니즘을 이해하기 위해 연구진은 생성된 서열의 잔기 패턴을 추가로 분석했습니다. 이전 연구에서는 트립토판(W), 페닐알라닌(F), 티로신(Y)으로 구성된 소수성 삼중체와 아르기닌(R), 라이신(K)과 같은 양전하를 띤 잔기가 세포 투과 펩타이드가 막 수송을 달성하는 데 중요한 특징이라는 것을 보여주었습니다.
분석 결과 다음과 같은 사실이 밝혀졌습니다.Bi-TEAM의 안내에 따라 생성된 서열에서 소수성 삼중체와 두 개의 양전하를 띤 잔기의 동시 발생 빈도가 크게 증가했으며, 잔기 수 분포 또한 일관된 경향을 보였다.이러한 농축 패턴은 막 투과 펩타이드의 알려진 구조-기능 규칙과 매우 일치하며, 이는 Bi-TEAM이 관련 생물학적 메커니즘을 포착할 뿐만 아니라 생성 과정에서 막 투과 특성을 가진 서열의 확률을 크게 높일 수 있음을 나타냅니다. 제어 변수 분석을 통해 펩타이드 길이(10~20개 잔기)의 영향이 배제되었으며, 이는 모델이 실제로 샘플링 분포를 막 수송에 더 유리한 화학-생물학적 공동 위치 공간으로 유도함을 보여줍니다.
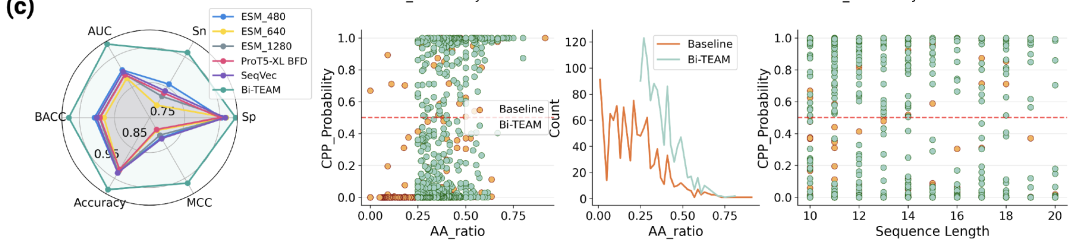
중간 부분: 주요 소수성 잔류물의 풍부도와 침투 확률 간의 관계;
오른쪽: 고리형 펩타이드 길이와 침투 확률 간의 관계
마지막으로, 본 연구는 사례 연구를 통해 구조적 수준에서 결과를 검증했습니다. 연구진은 먼저 아플리버셉트 이량체의 3차원 구조를 시각화하고 정전기적 전위에 따라 분자 표면에 색을 입혔습니다. 그런 다음, AlphaFold3를 사용하여 아플리버셉트와 고리형 펩타이드의 복합체 구조를 예측하고 설계했습니다. 분석 결과, 두 개의 잠재적인 고리형 펩타이드 결합 포켓이 확인되었습니다. 하나는 세 개의 고리로 구성된 소수성 공동이고, 다른 하나는 고리 구조와 β-시트 조각으로 형성된 포켓입니다. 이러한 구조 정보는 향후 고리형 펩타이드 최적화 및 잠재적인 임상 적용을 위한 중요한 기반을 제공합니다.

펩타이드 약물 개발 분야의 기술 혁신에 중점을 둡니다.
펩타이드 과학 분야에서는 기초 연구에서 임상 적용에 이르기까지 전 세계 수많은 연구 기관들이 주요 질병을 정복하기 위한 새로운 기술적 경로와 치료법을 적극적으로 모색하고 있습니다.
예를 들어, 영국 브리스톨 대학교 생화학과의 구조 생물학 연구팀이 있습니다.극저온 전자 현미경 및 X선 결정학 같은 첨단 기술을 사용하여 면역 체계의 미세 구조를 분석하고, 이를 기반으로 구조 기반 펩타이드 약물 설계를 수행합니다.그들은 인체 보체 시스템을 정확하게 활성화할 수 있는 고리형 펩타이드 분자를 설계함으로써 자가면역 질환 치료를 위한 차세대 약물 후보 물질을 개발하려고 시도하고 있습니다.
한편, 킹스 칼리지 런던과 자그레브 대학교의 협력 프로젝트인 톡시코드(ToxiCode)는 동물 독에서 새로운 약물을 발견하는 독특한 방법을 연구하고 있습니다.이 프로젝트는 인공지능과 합성생물학을 결합하여, 하이브리드 AI 시스템을 활용해 펩타이드 서열 패턴과 구조-활성 관계를 학습함으로써 암, 신경 질환 및 감염성 질환을 표적으로 하는 새로운 생리활성 펩타이드를 신속하게 설계할 수 있도록 합니다.이는 지속 가능하고 윤리적인 신약 개발을 위한 새로운 방법론적 틀을 제공합니다.
이는 펩타이드 신약 개발이 점차 새로운 연구 패러다임을 형성하고 있음을 보여줍니다. 구조 생물학, 인공지능, 화학 생물학이 융합되면서 기초 연구와 산업 개발의 경계가 모호해지고 있습니다. 새로운 분자들은 종종 여러 학문 분야의 기술적 결합을 통해 탄생하지만, 임상 적용 가능성을 진정으로 결정하는 것은 실험실 발견에서 산업화에 이르는 점진적으로 확립되는 중개 경로입니다. 이러한 과정에서 펩타이드 분자는 큰 분자와 작은 분자 사이의 독특한 특성 덕분에 재평가되고 있으며, 점점 더 많은 질병 분야에서 새로운 응용 가능성을 보여주고 있습니다.
참조 링크:
1.https://www.bristol.ac.uk/news/2025/november/bristol-researcher-awarded-over-850000-to-develop-new-treatments.html
2.https://www.kcl.ac.uk/news/kings-to-collaborate-in-venom-based-drug-discovery-project
3.https://mp.weixin.qq.com/s/X67D1qrUzclwOsJ9cKUtZg








