Command Palette
Search for a command to run...
코넬 대학교는 고전도성 리튬 이온 전해질의 화학적 메커니즘을 해독하는 혁신적인 AI 프레임워크를 제안했으며, %에 대해 80% 이상의 예측 성공률을 달성했습니다.

신에너지 배터리 시장이 급속도로 확장됨에 따라, 특히 리튬이온 배터리, 고체 배터리 및 고에너지 밀도 배터리의 광범위한 적용으로 인해 전해질 성능 최적화는 배터리의 안전성, 효율성 및 수명을 결정하는 핵심 요소가 되었습니다.
염-용매 화학은 대부분의 리튬 이온 배터리 시스템에서 전해질의 거동을 뒷받침하며, 이온 전도도, 점도, 화학적 안정성과 같은 주요 특성을 결정합니다. 그러나 전해질의 합리적인 설계는 무수히 많은 조합과 비선형적인 구조-성능 상호 작용을 포함하는 방대한 화학적 공간으로 인해 제약을 받습니다. 희소하고 불균등하게 분포된 실험 데이터는 이러한 문제를 더욱 악화시켜 모델의 일반화 능력을 저해합니다. 최근 몇 년 동안 AI 기반 자율 전해질 탐색이 어느 정도 진전을 이루었지만,그러나 기존 연구는 대규모 전해질 조성물이 포괄하는 방대한 화학적 공간을 탐색하면서 본질적인 해석 가능성을 유지할 수 있는 통합된 계산 패러다임이 여전히 부족하다는 점을 분명히 보여줍니다.
이러한 맥락에서,코넬 대학교의 연구팀이 염-용매 화학을 모델링하고 해석하기 위한 견고하고 해석 가능하며 데이터 효율적인 프레임워크인 SCAN을 개발했습니다.이 프레임워크는 긴 꼬리 분포 데이터를 효과적으로 처리하고 염-용매 조성의 전체 스펙트럼을 포착합니다. 연구진은 SCAN을 비수용성 전해질(NAE) 시스템에 적용하여 전도도 예측에서 0.372 mS·cm⁻¹의 기준 오차를 달성했으며, 기준 모델 대비 예측 오차를 65.31 TP³T 감소시켰습니다.
더욱 중요한 것은 대규모 검증을 통해 다음과 같은 사실이 밝혀졌다는 것입니다.해당 모델은 최상위 후보 시스템에 대해 81.08%의 예측 성공률을 달성했습니다.SCAN은 예측 능력 외에도 기울기 분리, 기호 회귀 및 양자 화학 계산을 도입하여 분자 유연성과 이온-용매 상호 작용이 전도도에 미치는 영향을 밝혀냄으로써 화학적 메커니즘을 규명합니다.
"염-용매 화학을 위한 동적 경로 안내형 해석 가능 프레임워크"라는 제목의 관련 연구 결과가 Nature Computational Science에 게재되었습니다.
연구 하이라이트:
* SCAN은 고성능 NAE 염-용매 화학 연구의 핵심적인 공백을 메웁니다.
원자 중심의 잠재 에너지 표면 모델에서 영감을 받아, 우리는 특징 중심의 전용 표현과 어텐션 메커니즘을 갖춘 다중 특징 네트워크(MFNet)를 개발했습니다.
* 혁신적으로, MFNet에는 동적 라우팅 전략이 도입되어 원본 데이터 분포를 변경하지 않고도 넓은 범위에 걸쳐 이온 전도도를 정확하게 예측할 수 있습니다.

서류 주소:
https://www.nature.com/articles/s43588-026-00955-5
저희 공식 위챗 계정을 팔로우하고 백그라운드에서 "스캔"이라고 답장하시면 전체 PDF 파일을 받으실 수 있습니다.
데이터 차원은 광범위하게 다루어집니다.
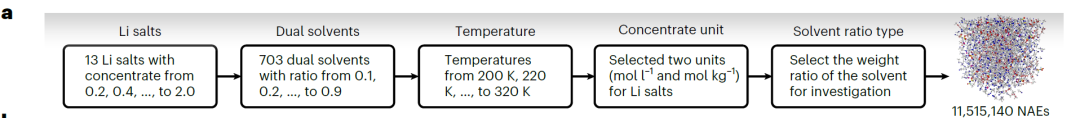
고정밀 SCAN 모델을 학습시키기 위해,연구팀은 13가지 리튬염과 38가지 유기용매(아래 그림 참조)를 포함하는 총 13,302개의 완전한 데이터 항목으로 구성된 CALiSol 데이터 세트를 구축했습니다.
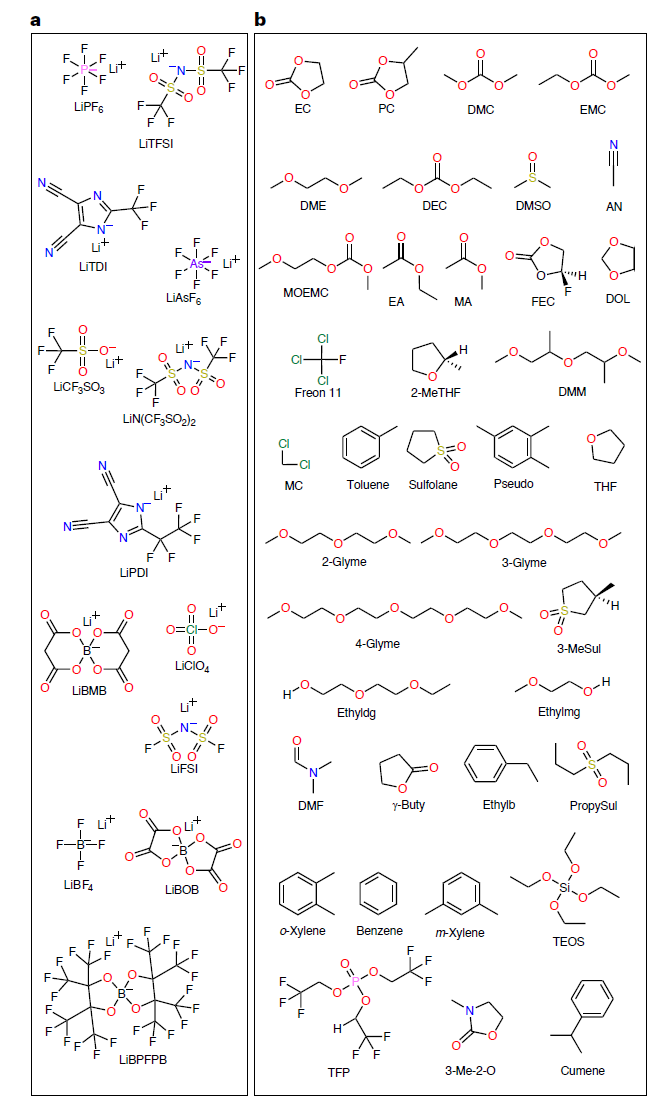
데이터의 차원은 매우 광범위하며, 각 데이터 포인트에는 다음 내용이 포함됩니다.
* 이온 전도도 k: 0–38.1 mS/cm
* 온도 T: 194.15–477.42 K
* 염 농도 c: 0–4 mol/L 또는 mol/kg
* 리튬염 종류: LiPF₆, LiTFSI, LiFSI, LiBOB 등 총 13가지 종류.
* 용매 종류: 에틸렌 카보네이트(EC), 메틸 에틸 카보네이트(EMC), 프로피오니트릴(AN) 등을 포함한 다양한 용매.
* 혼합 전략 SRT: 몰비, 부피비 또는 질량비
모든 리튬 염과 용매의 분자 정보는 SMILES 시퀀스를 사용하여 3차원 분자 좌표로 변환되었고, 정확하고 신뢰할 수 있는 분자 구조 및 전자적 특성을 확보하기 위해 B3LYP/6-31G 이론 수준에서 기하 최적화가 수행되었습니다. 이 방법을 통해,이 데이터 세트는 이온 전도도 값뿐만 아니라 각 시스템에 대한 분자 특성, 구조 정보 및 용매화 환경에 대한 정보도 제공합니다.데이터의 무결성과 과학적 해석 가능성 사이의 균형을 유지하면서 AI 모델에 풍부한 입력값을 제공합니다.
데이터셋 구축 과정에서 연구팀은 긴 꼬리 분포(LTD) 문제에 특별히 주의를 기울였습니다. 즉, 전도성이 높은 시스템의 수는 제한적인 반면, 대부분의 시스템의 전도성은 낮습니다. 9,115개의 NAE(약 68.5%)의 경우 k < 5 mS·cm⁻¹이고, 67개(약 0.5%)에 대해서만 k > 20 mS·cm⁻¹입니다.
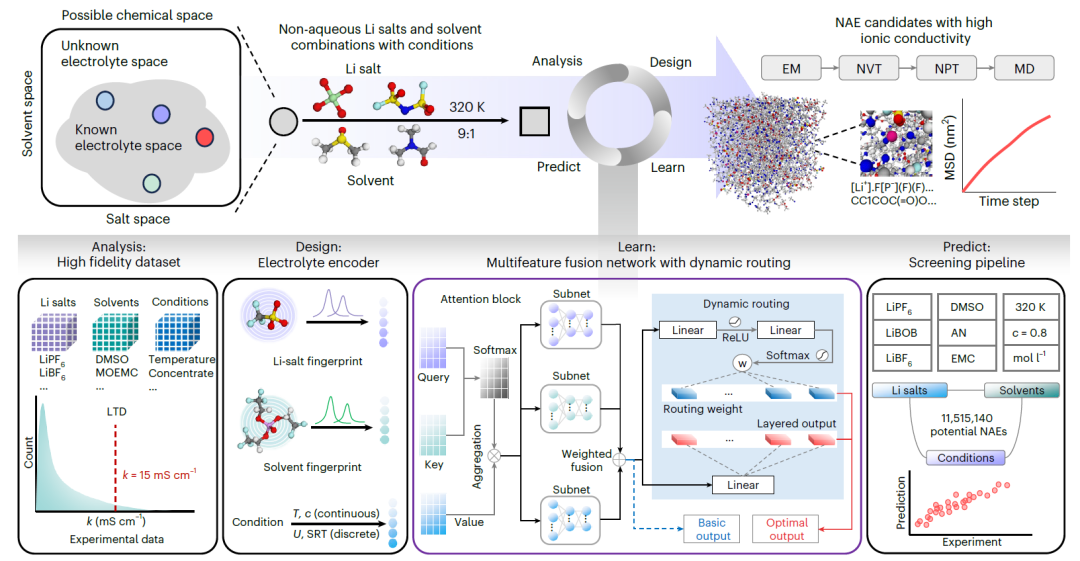
SCAN: MFNet 및 동적 라우팅 메커니즘을 사용합니다.
SCAN은 동적 라우팅을 기반으로 하는 NAE 엔지니어링 플랫폼입니다. 다음 다이어그램은 전체 워크플로우를 보여줍니다.
분석
리튬염, 용매 및 실험 조건의 다양한 조합을 포괄하는 고충실도 실험 데이터 세트를 구축했습니다. 각 변수에 대해 통계 분석을 수행했으며, 여기에는 k 값의 긴 꼬리 분포 특성 분석이 포함되었습니다.
설계
리튬염과 용매에 대해 각각 화학 정보를 포함하는 분자 지문을 설계하고, 이를 조건부 코딩과 결합하여 염-용매-조건 조합을 정확하게 특성화했습니다.
배우다
본 연구에서는 NAE의 k 값을 예측하기 위해 맞춤형 다중 특징 융합 네트워크(MFNet)를 개발했습니다. 염, 용매, 조건의 표현을 각각 처리하기 위해 쿼리, 키, 값 데이터를 임베딩하여 독립적인 어텐션 모듈을 구축한 후, 완전 연결 신경망을 이용하여 융합했습니다. 또한, 모델 성능과 견고성을 향상시키기 위해 동적 라우팅 전략을 도입했습니다.
예측하다
천만 개 이상의 염-용매 조합에서 잠재적인 NAE를 식별하기 위해 고처리량 스크리닝 파이프라인을 구축했습니다. 예측된 k 값이 높은 후보 시스템은 에너지 최소화(EM), 정준 앙상블(NVT), 등온-등압 앙상블(NPT), MD 생산 시뮬레이션, 평균 제곱 변위(MSD) 분석 및 방사형 분포 함수(RDF) 계산을 포함한 분자 동역학(MD) 시뮬레이션을 사용하여 추가적으로 검증했습니다.
MFNet과 동적 라우팅 메커니즘은 전체 프레임워크의 핵심이며, 구체적인 메커니즘은 다음과 같습니다.
MFNet: 멀티채널 셀프 어텐션 네트워크
원자 중심 전위 표면 모델에서 영감을 받은 MFNet(분자 특징 네트워크) 프레임워크는 네트워크를 각각 다른 기능을 처리하는 세 개의 독립적인 하위 네트워크로 나눕니다.
* 리튬염 서브넷: CALiSol 데이터 세트에서 각 NAE는 한 가지 유형의 리튬염만 포함하므로 해당 설명자 벡터가 리튬염 서브넷에 직접 입력됩니다.
* 용매 하위 네트워크: 일부 데이터 포인트는 여러 용매를 포함하므로(예: PC와 AN을 0.9:0.1 비율로 혼합), 전체 용매 환경을 반영하기 위해 이러한 용매 특성값의 평균값을 입력값으로 계산합니다.
* 조건부 서브넷: 온도 및 농도와 같은 실험 조건을 처리합니다.
셀프 어텐션 모듈 다음에는 두 개의 완전 연결 은닉층이 사용되어 입력에 대한 점진적 투영 및 비선형 변환을 수행하고, 이를 통해 고차원 특징을 생성하여 후속 처리의 표현력을 향상시킵니다. 각 층 사이에는 ReLU 비선형 활성화 함수를 사용하여 모델의 표현력을 더욱 향상시킵니다.
최종 네트워크 아키텍처는 리튬염 서브넷(14–16–16), 용매 서브넷(14–16–16), 조건부 서브넷(6–16–16)으로 구성됩니다. 고유 차원(특징 차원 <128)을 갖는 잠재적 출력은 가중 융합 모듈로 입력되어 입력 특징의 전역 의존성 정보를 유지하면서 가중 출력을 계산합니다. 리튬염, 용매, 조건부 특징의 인코딩은 세 개의 서브넷에서 독립적으로 처리되므로, 단일 헤드 셀프 어텐션(SHA)을 사용하여 소규모 특징 데이터(각각 14, 14, 6차원)를 효율적으로 처리할 수 있어 임베딩 차원이 낮은 작업에 적합합니다.
동적 라우팅 메커니즘: 롱테일 데이터 문제 해결
긴 꼬리 분포에서 데이터 샘플은 주로 "머리 부분"에 집중되어 있고 "꼬리 부분"의 샘플은 드뭅니다. 그러나 기존 모델은 종종 머리 부분 샘플에 과적합되어 꼬리 부분을 무시하므로 중요하지만 부족한 화학적 공간을 충분히 탐색하지 못합니다. 이러한 문제를 해결하기 위해 본 연구에서는 MFNet에 동적 라우팅 전략을 도입합니다. 모든 샘플을 동일하게 처리하는 표준 아키텍처와 달리,동적 라우팅은 레이어 입력에 따라 서로 다른 레이어에 서로 다른 표현 기능을 적응적으로 할당하는 소프트 게이팅 메커니즘을 학습합니다.아래와 같이 표시됩니다.

이 메커니즘은 희귀 샘플이 서로 다른 라우팅 경로를 활성화하고 조건부 계산을 용이하게 하여 빈도가 낮은 범주에 대한 일반화 능력을 향상시킵니다. 이 메커니즘의 두 가지 핵심 특징은 다음과 같습니다.
* 입력 종속 라우팅 가중치: 즉, 원래 손실 함수나 데이터 분포를 수정하지 않고 각 샘플에 대해 특징 부분 공간을 선택하는 것.
* 범주 적응형 기능 분리: 이는 지배적인 범주와 꼬리 범주 간의 차이를 명시적으로 모델링하는 것을 포함하며, 동적 라우팅이 단순한 정적 손실 재구성보다 더 유연하고 해석 가능한 LTD 솔루션을 제공한다는 것을 보여줍니다.
해석 가능성: GBA와 기호 회귀
SCAN은 이온 전도도를 정확하게 예측하는 동시에 화학적 해석 가능성을 제공합니다.
* GBA(그라디언트 기반 기여도 분석):SCAN 모델은 3개의 병렬 어텐션 신경망 프레임워크를 사용하기 때문에 트리 모델에 비해 의사 결정 과정을 시각화하기가 더 복잡합니다. k에 영향을 미치는 주요 화학적 요인을 파악하기 위해 GBA 방법을 사용하여 특징 중요도를 평가했습니다. 즉, 각 입력 특징이 모델 출력에 미치는 기울기 기여도를 계산하고 가장 중요한 리튬 염, 용매 및 조건부 특징을 식별했습니다.
* 기호 회귀 분석:핵심 리튬 염, 용매 및 조건 정보와 kkk 사이의 해석 가능한 함수적 관계를 밝히기 위해 PySRRegressor 기반의 기호 회귀 분석 방법을 사용했습니다.
SCAN 모델은 모든 기준 모델보다 일관되게 우수한 성능을 보입니다.
MFNet과 동적 라우팅을 사용하는 SCAN 프레임워크의 성능을 종합적으로 평가하기 위해 연구진은 MFNet 기반의 기준 모델 4개를 구축했습니다.이들 중 어느 것도 동적 라우팅을 포함하지 않습니다: (1) 기본 MFNet 모델: B1(처리되지 않은 디스크립터) 및 B2(최대값으로 스케일링된 설명자); (2) 소수 클래스 과표집 기법 SMOTE를 결합한 MFNet 모델: S1-에스18(3) K-최근접 이웃 언더샘플링 기법(KUTE)을 결합한 MFNet 모델: U1-유9(4) SMOTE와 KUTE를 결합한 MFNet 모델: SU1–SU9 .
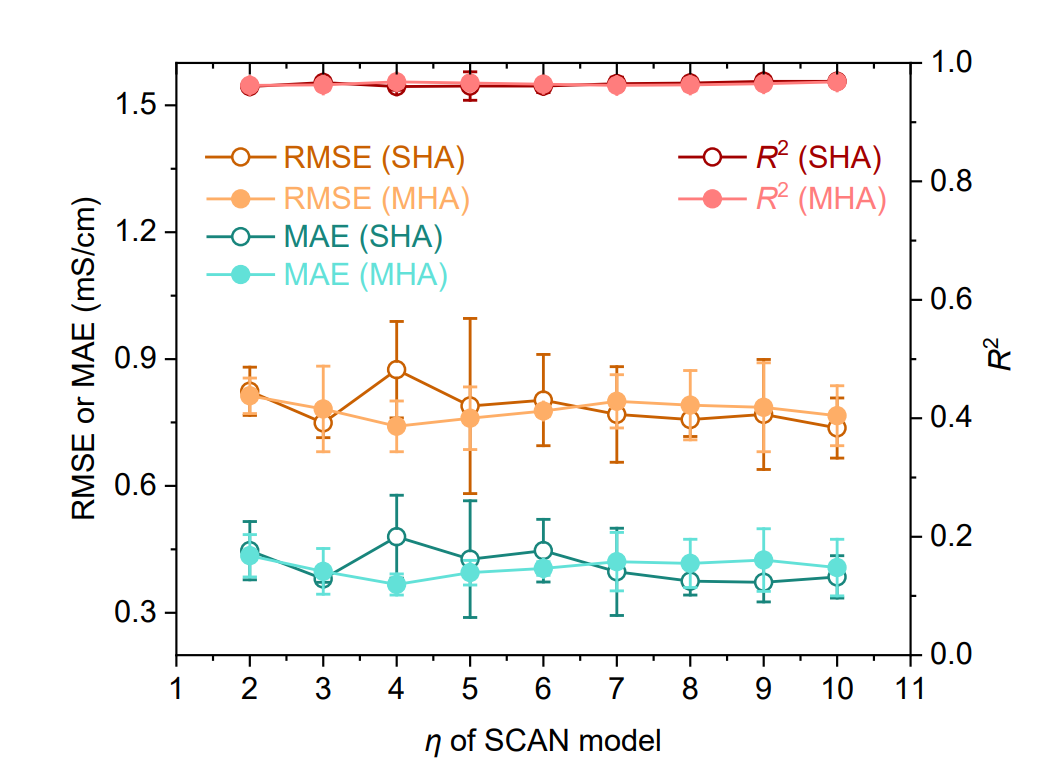
동시에, 아래 그림에서와 같이 Transformer 기반 모델, 그래프 신경망(GNN), 서포트 벡터 회귀(SVR), 그리고 익스트림 그래디언트 부스팅(XGBoost)을 기준 비교 모델로 사용했습니다. 또한, 모델의 견고성을 검증하기 위해 단일 헤드 어텐션(SHA) 또는 다중 헤드 어텐션(MHA) 구조와 다양한 개수의 동적 라우팅 레이어(η = 2–10)를 사용하는 SCAN 모델을 구현했습니다.
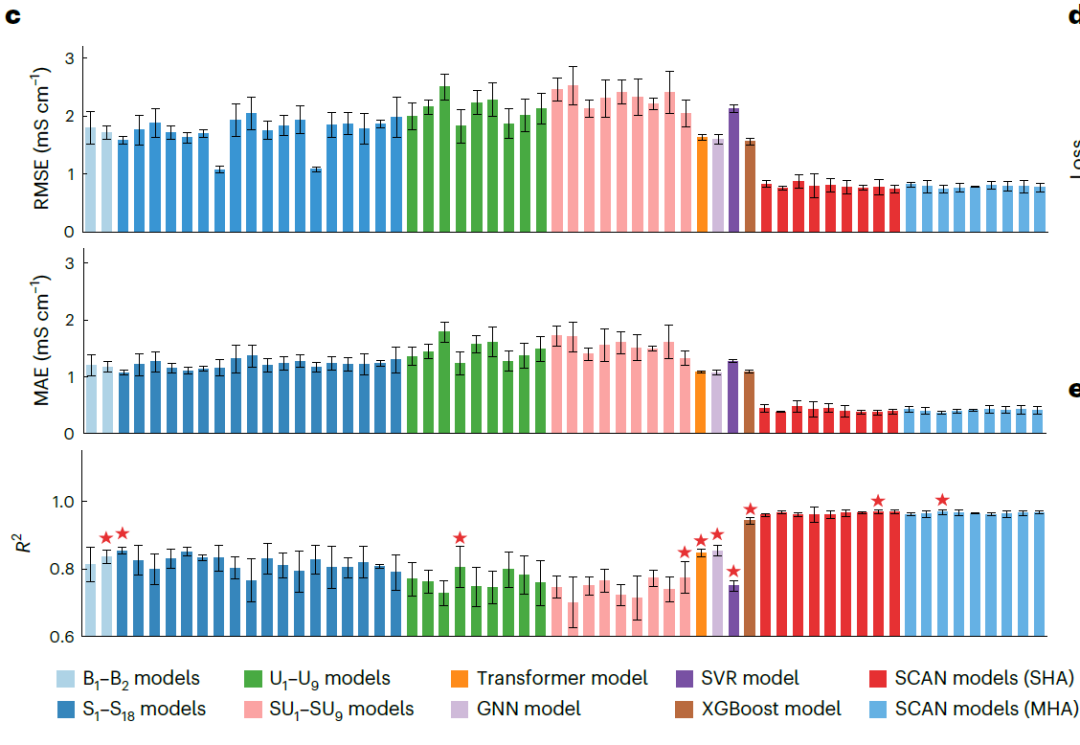
NAE 성능 평가
아래 그림에 나타난 결과는 SHA(η = 9)를 사용한 SCAN 모델이 모든 기준 모델보다 일관되게 우수한 성능을 보이며, 예측 오차가 현저히 낮고(RMSE 0.769 mS·cm⁻¹, MAE 0.372 mS·cm⁻¹) R² 값이 더 높다(0.969)는 것을 보여줍니다. 특히 주목할 만한 점은...MAE에서 가장 우수한 성능을 보인 기준 GNN(1.072 mS·cm⁻¹)과 비교했을 때, 오차는 65.31 TP3T만큼 감소했습니다.MHA(η = 4)를 사용한 SCAN 성능은 SHA 버전과 유사하며, 이는 MFNet이 동적 라우팅과의 통합에서 우수한 견고성을 가지고 있음을 나타냅니다.
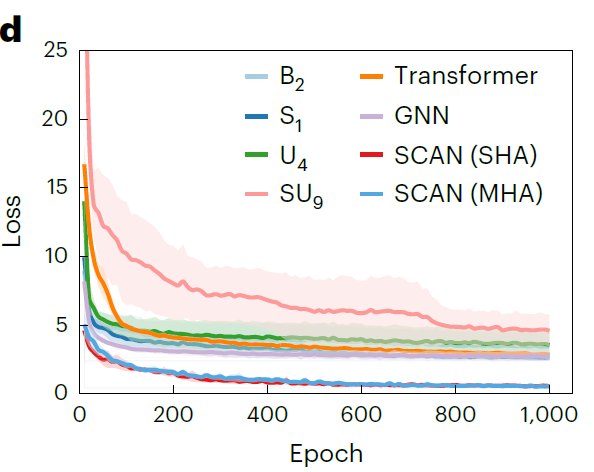
또한,연구진은 1,000번의 훈련 에포크에 걸쳐 SCAN과 대표적인 기준 모델의 검증 손실 곡선을 그렸습니다.아래 그림에서 볼 수 있듯이, SCAN의 예측 오차는 학습 초기부터 기준 모델보다 현저히 낮으며, 초기 손실은 4.59 mS·cm⁻¹에 불과하고, 검증 손실은 지속적으로 감소하여 더욱 낮은 수준으로 수렴합니다. 이는 SCAN이 더 강력한 안정성과 일반화 능력을 가지고 있음을 보여줍니다.
고처리량 NAE 스크리닝 기능
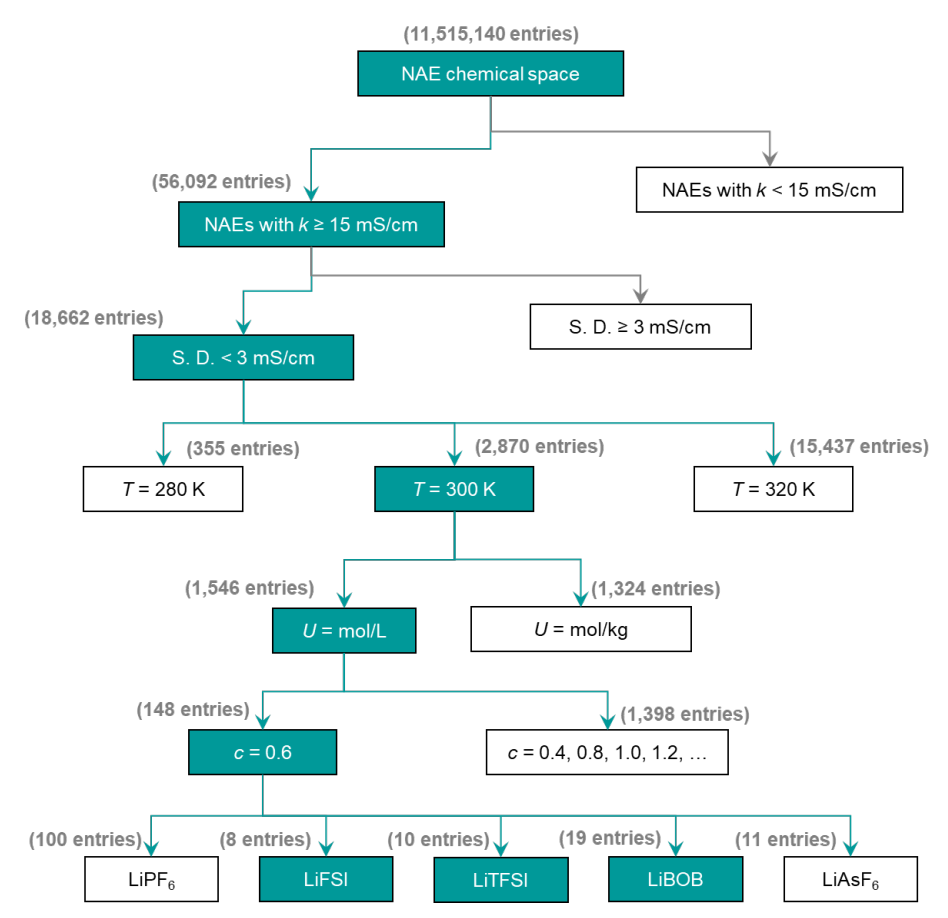
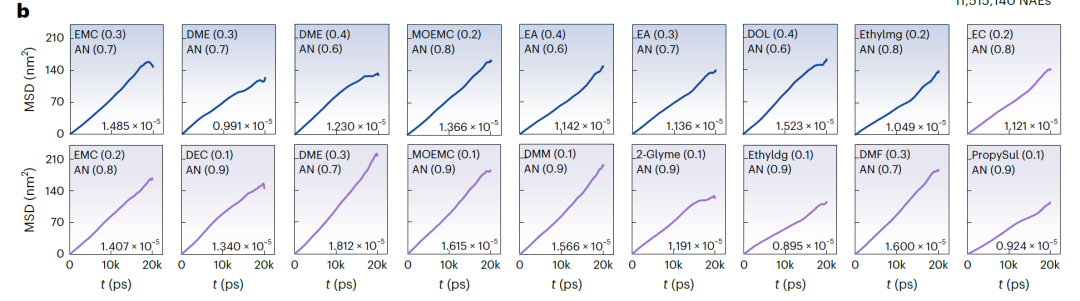
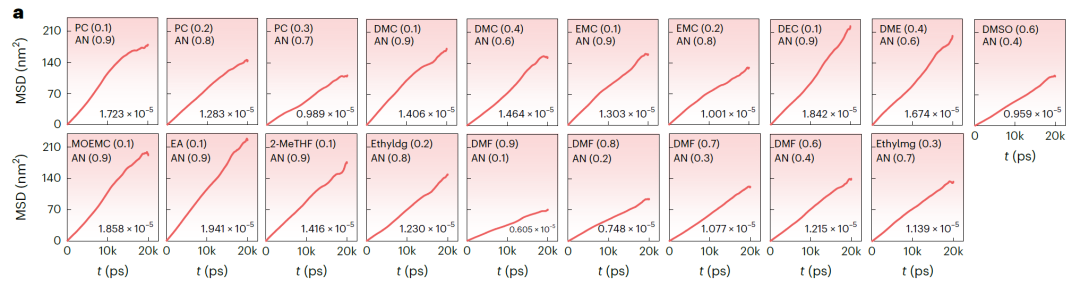
고정밀 스캐닝은 NAE의 확장된 화학적 공간을 효율적으로 탐색할 수 있습니다. 이를 위해 연구진은 높은 k 값을 갖는 NAE를 식별하도록 설계된 고처리량 스크리닝 프로세스(아래 그림 참조)를 개발했습니다.이를 바탕으로 11,515,140개의 잠재적 이중 용매 NAE가 생성되었습니다.
SCAN의 예측력을 활용하여 k ≥ 15 mS·cm⁻¹인 56,092개의 NAE를 신속하게 식별했으며, 그중 18,662개는 예측 불확실성이 낮았습니다(<3 mS·cm⁻¹). 이 과정을 통해 분자 동역학 시뮬레이션 및 실험의 계산 및 비용 부담을 크게 줄일 수 있습니다.
비수용성 전해질(NAE) 후보 물질의 검증
연구진은 예측 결과를 엄밀하게 검증하기 위해 분자 동역학(MD) 시뮬레이션을 수행하여 k 값을 얻었습니다. 온도(T), 농도(c) 및 실제 조건을 기반으로 MD 검증 범위를 LiFSI, LiTFSI 및 LiBOB 기반의 NAE 후보 시스템으로 좁혔고, 최종적으로 아래 그림과 같이 상세 연구를 위해 37개의 유망한 시스템을 선정했습니다.
각 시스템의 평균 계산 시간은 약 10시간(36,355.12초)이며, 10⁷개의 후보 시스템을 시뮬레이션하는 데 드는 예상 비용은 약 10⁸ GPU 시간으로, 이상적인 고성능 컴퓨팅 환경에서도 무차별 대입 방식의 방법론적 선택으로는 현실적으로 불가능한 수준입니다. 이에 비해,잘 훈련된 SCAN 모델은 각 후보 시스템에 대해 설명자 계산부터 최종 예측까지 전체 프로세스를 5초 이내에 완료할 수 있어 계산 비용을 7,200배 이상 절감할 수 있습니다.이는 확장성과 효율성을 크게 향상시킵니다. 이러한 점은 NAE 발굴에서 SCAN 프레임워크의 필요성을 강조하며, 해당 프레임워크의 대리 모델은 매우 낮은 계산 비용으로 고성능 후보를 신속하게 우선순위화할 수 있습니다.
아래 그림 b(LiFSI 및 LiTFSI 기반 NAE)와 그림 a(LiBOB 기반 NAE)는 시간에 따른 평균 제곱 변위(MSD)와 MD 시뮬레이션에서 얻은 해당 확산 계수를 보여줍니다. 모든 시스템의 MSD는 안정적인 선형 증가를 나타내어 시뮬레이션이 안정적이고 수렴함을 보여줍니다. SCAN으로 예측된 k 값은 MD 시뮬레이션 결과와 매우 일치하며, 평균 편차는 3.198 mS·cm⁻¹에 불과하고 최대 편차는 7.342 mS·cm⁻¹입니다. 검증 시스템에서 k > 15 mS·cm⁻¹인 25개 시스템의 경우 검증 성공률은 67.571TP⁻¹였습니다. 예측된 k > 14 mS·cm⁻¹인 시스템의 경우 검증 성공률은 81.081TP⁻¹로 증가했습니다.
인공지능은 배터리 연구 개발의 근본적인 패러다임을 바꾸고 있습니다.
배터리는 가전제품, 전기 자동차, 전력망 에너지 저장 시스템 등 주요 응용 분야의 핵심 구성 요소로서 전 세계적인 지속 가능한 에너지 전환을 주도하고 있습니다. 에너지 및 전력 밀도 향상, 수명 연장, 안전성 강화, 제조 비용 절감은 배터리 연구 개발의 핵심 목표입니다. 이러한 목표를 달성하는 열쇠는 배터리 내부의 전기화학적 메커니즘, 특히 전기화학적 계면 반응 및 안정화 메커니즘, 전자와 이온의 결합 및 이동, 그리고 차세대 전극 소재의 에너지 저장 메커니즘에 대한 심층적인 이해에 있습니다.
기술 발전의 더 넓은 관점에서 볼 때, SCAN 프레임워크로 대표되는 "데이터 기반 + 해석 가능한 모델링" 패러다임은 차세대 배터리 연구 개발 시스템의 중요한 구성 요소가 되고 있습니다. 지난 수십 년간 배터리 소재 시스템의 혁신은 주로 경험에 기반한 시행착오 실험에 의존해 왔으며, 이로 인해 개발 주기가 길어지고 비용이 많이 들었습니다. 그러나 머신러닝과 고성능 컴퓨팅의 통합으로 배터리 연구 개발은 점점 더 효율적으로 이루어지고 있습니다.
예를 들어,바이트댄스의 시드 팀 소속인 웬 얀과 셩 곤을 비롯한 연구진은 전방 예측 모델과 역방향 생성 방법을 통합한 전해질 제형 설계 통합 프레임워크를 개발했습니다.연구진은 속성 태그가 부착된 단일 분자(24만 개 이상) 및 분자 혼합물(1만 개 이상)에 대한 광범위한 문헌 자료를 수집하여 전해질 설계 공간을 포괄적으로 다루었습니다. 여기에 분자 동역학 시뮬레이션을 통해 얻은 10만 개 이상의 분자 혼합물 데이터를 추가로 활용하여, 리튬 금속 배터리의 계면 안정성과 관련된 용융 구조뿐만 아니라 전도도까지 예측할 수 있는 정확한 머신러닝 모델을 개발했습니다.
논문 제목: 액체 전해질 조성에 대한 통합 예측 및 생성 솔루션
논문 링크:
https://www.nature.com/articles/s42256-025-01173-w
전반적으로 인공지능은 배터리 연구 개발의 근본적인 패러다임을 재편하고 있습니다. 모델과 실험 시스템의 심층적인 통합을 통해 배터리 소재의 발견 속도와 혁신 밀도가 새로운 가속 주기에 진입할 것으로 기대됩니다.
참고문헌:
https://www.nature.com/articles/s43588-026-00955-5
https://phys.org/news/2026-02-ai-framework-reveals-chemistry-high.html
https://static-content.springer.com/esm/art%3A10.1038%2Fs43588-026-00955-5/MediaObjects/43588_2026_955_MOESM1_ESM.pdf
https://www.eet-china.com/mp/a471613.html