Command Palette
Search for a command to run...
10万通りの可能性から合成の成功まで、グローバルアテンションメカニズムとAIモデルCGformerの革新的な統合により、高エントロピー材料の開発が容易になります。

人工知能は材料科学研究開発のパラダイムを大きく変革し、新材料の発見を加速し、その性能を最適化する上で画期的な価値を発揮しています。高スループットコンピューティングと機械学習の緊密な統合により、長い実験サイクルや膨大なリソース消費といった従来の「試行錯誤」手法の問題点が効果的に解決されています。材料探索は、「計算駆動型実験検証」の効率的な反復段階に入りました。しかし、人類の技術やライフスタイルの革新に伴い、新エネルギーや航空宇宙などの分野における新素材に対する性能要件はますます厳しくなり、特に高エントロピー材料の研究開発の分野では、従来の機械学習手法の限界が徐々に明らかになりつつあります。
高エントロピー材料は、複数の主要元素を混合して作られる新しいタイプの材料です。これらの元素の相乗作用により、高エントロピー材料は原子配列の配置エントロピー(すなわち無秩序性)を大幅に増加させ、従来の材料と比較して優れた機械的特性、高温特性、耐腐食性を実現します。これらの材料は、エネルギー貯蔵、航空宇宙、極限環境機器への応用に大きな可能性を秘めています。
クリスタル グラフ畳み込みニューラル ネットワーク (CGCNN) やアトミスティック ライン グラフ ニューラル ネットワーク (ALIGNN) などの従来のアプローチにはすべて、アーキテクチャ上の欠陥があります。局所的な情報相互作用メカニズムによって制限されるため、長距離の原子相乗効果をモデル化することが難しく、複雑な結晶構造に特有のグローバル情報を完全に捕捉することができず、予測精度が制限されます。同時に、高エントロピー材料の固有の特性により、その研究開発は従来の材料をはるかに超える課題に直面しています。複雑な微細構造、高品質の実験データの不足、そして動的に無秩序な原子挙動が、高エントロピー材料の開発における主な障害となっています。
ツールの欠陥やアップグレードの要求に応じて、上海交通大学人工知能・微細構造研究室(AIMS-Lab)の李金金教授と黄福強教授のチームは、従来のモデルの限界を打ち破る新しいAI材料設計モデルCGformerを開発しました。このモデルは、GraphormerのグローバルアテンションメカニズムとCGCNNを革新的に組み合わせ、中心性コーディングと空間コーディングを統合することで、結晶図を通じて物質構造を直感的に記述できるだけでなく、「グローバルアテンション」メカニズムの助けを借りて長距離原子間の相互作用を捉え、従来の「隣接原子のみに焦点を当てる」モデルにはないグローバル情報処理機能を獲得しました。
この手法は、より包括的な構造情報を提供することで、構造内のイオン移動をより正確に予測することを可能にし、特に高エントロピーで複雑な結晶性材料を含む新材料の研究開発に信頼性の高いツールを提供します。この研究成果は、「CGformer:材料特性予測のためのグローバルな注目を集めるTransformer強化結晶グラフネットワーク」というタイトルで、主要学術誌Matterに掲載されました。
研究のハイライト:
* グローバルアテンションメカニズムに基づく AI 材料設計モデルである CGformer の研究開発は、材料研究開発科学に信頼性が高く強力なツールを提供し、複雑な結晶構造の発見プロセスを加速するのに役立ちます。
* CGCNNと比較して、CGformerは高エントロピーナトリウムイオン固体電解質(HE-NSE)の研究において平均絶対誤差が25%減少し、その実用性と進歩性を効果的に実証しました。
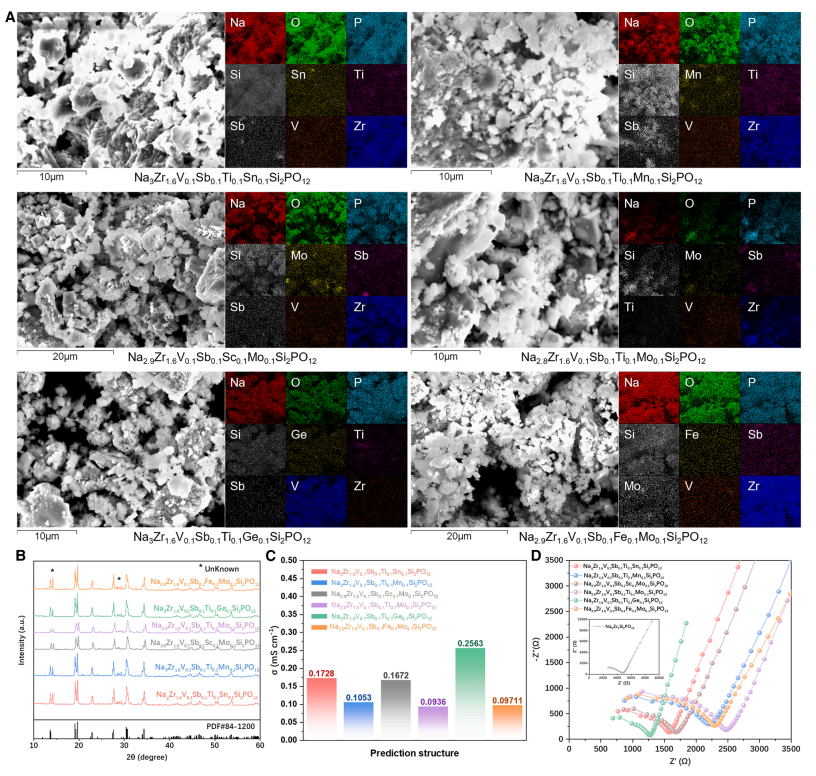
*148,995個の高エントロピー構造の中から18個の可能性のあるものを選別し、室温でのナトリウムイオン伝導率が0.256 mS/cmに達する6個の高エントロピーナトリウムイオン固体電解質(HE-NSE)の合成と検証に成功し、その実用価値を実証しました。

用紙のアドレス:
https://www.cell.com/matter/abstract/S2590-2385(25)00423-0
公式アカウントをフォローし、「CGformer」と返信すると、完全なPDFが手に入ります。
AIフロンティアに関するその他の論文:
マルチカテゴリデータセットはCGformerモデルの機能を向上します
この研究の目的は、事例ベースのソリューションを通じて、高エントロピーシステムにおけるデータ不足と構造の複雑さによってもたらされる課題に対処することです。この研究ケースは、新エネルギー電気自動車とグリッドエネルギー貯蔵アプリケーションに焦点を当てており、特に高エントロピーナトリウムイオン固体電解質の性能予測とスクリーニングに焦点を当てています。CGformerモデルのトレーニング、微調整、実験検証をサポートするために、以下のように様々なデータセットが構築され、使用されました。
ナトリウムイオン拡散エネルギー障壁(Eb)基本データセット:これは、本研究のために研究者らが構築した高エントロピー構造におけるナトリウムイオン拡散障壁に関する既知最大のデータセットです。ボロノイ分解による結晶構造解析(CAVD)法と結合価数サイトエネルギー(BVSE)法に基づいています。このデータセットは主にCGformerの事前学習に使用され、モデルがナトリウム含有構造に関連するグラフ情報を学習できるようにします。この情報は、計算された高エントロピーデータセットに転送され、高エントロピー材料のEb予測の基礎となります。
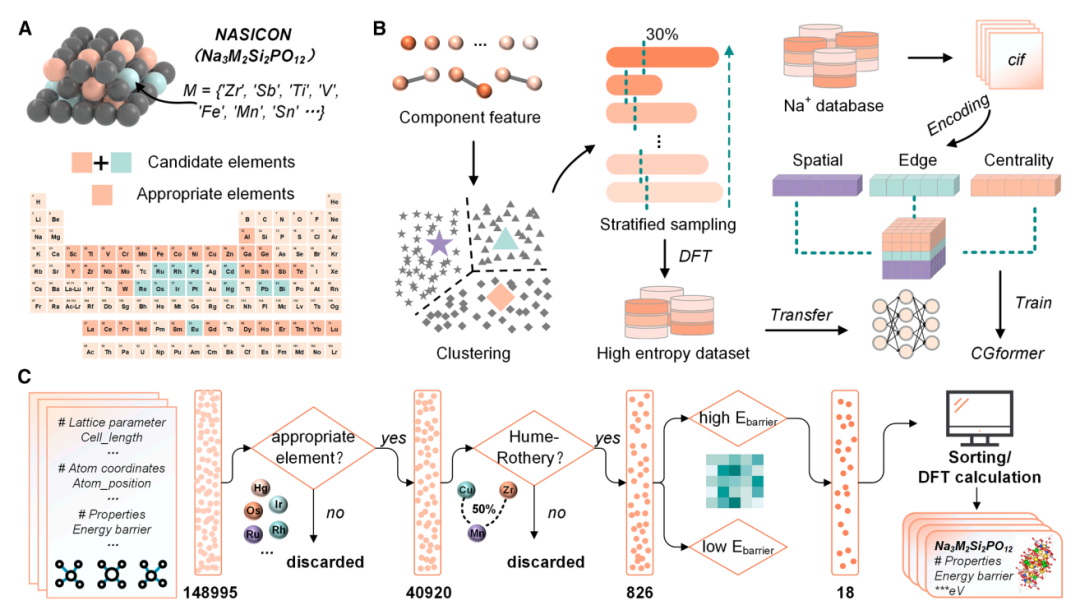
HE-NSEs計算データセット:Na₃Zr₂Si₂PO₁₂(上記参照)に基づき、Zrサイトに45種類の高エントロピードーピング元素が考慮され、148,995種類の高エントロピー構造を含む初期化学空間が構築されました。その後、不適格元素(放射性元素、高毒性元素、高価な元素)の除外、原子半径の差と電荷バランスの制約を含む複数回のスクリーニングを経て、化学空間はさらに826種類の比較的安定した構造に絞り込まれました。次に、教師なし階層的クラスタリングを用いて、これらの構造を20のグループに分類しました。各グループから301個のTP3T構造(合計238個)が階層的にサンプリングされ、密度汎関数理論(DFT)を用いてEb値が計算されました。これにより、最終的に CGformer を微調整するための専用データセットが形成され、特に高エントロピー NASICON 構造におけるナトリウムイオンの Eb を予測するタスクにモデルが適応され、対象シナリオにおけるモデルの精度が向上しました。
熱安定性評価データセット:研究者らは、Materials Projectデータベースから、凸包値(Ehull)を超えるエネルギーを持つすべてのナトリウム含有構造を抽出し、専用のトレーニングセットにまとめました。このデータセットは主に、HE-NSEの熱力学的安定性を評価するための補完モデルのトレーニングに使用されます。CGformerによって予測されたEbと組み合わせることで、このモデルは高性能と安定性の両方を備えた候補材料のスクリーニングに使用できます。
革新的な融合アーキテクチャがCGformerの「グローバル認識」を可能にします
CGformer は、従来の方法の欠点を解決するために根本的な革新を行い、2 つの高度なテクノロジーを統合して相補的な利点を実現しました。その中核は、結晶構造のグラフィカルな表現能力を保持し、グローバルアテンションメカニズムを通じて局所的な原子相互作用のみに焦点を当てるという制限を打ち破ることです。具体的には、Graphormer のグローバル アテンション メカニズムと CGCNN のクリスタル グラフ表現方法を組み合わせ、主要なエンコーディング モジュールを追加して、新しい情報処理パイプラインを構築します。
下の図 a は結晶図のエンコードプロセスを示しています。このプロセスは、実際の 3 次元結晶構造を、モデルで処理できる結晶図に変換することです。結晶構造中の原子はノードとして、原子間の化学結合はエッジとして表されます。この変換プロセスを通じて、研究者はノードとエッジの特徴、例えば様々な元素特性、電荷、共有結合半径、原子間距離、結合の種類、結晶の対称性情報などを抽出することができました。これらの特徴を組み合わせることで、CGformerに必要な初期入力データが得られ、結晶の化学情報と構造情報が完全に保持されました。
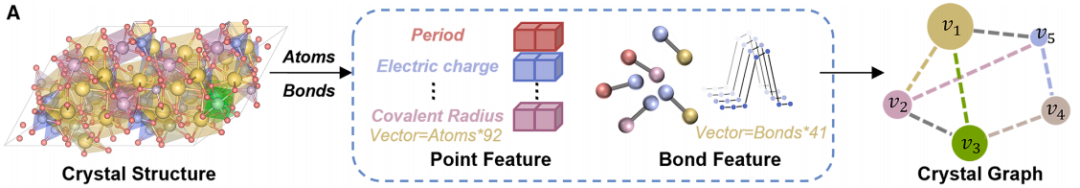
下の図bはCGformerのネットワークアーキテクチャを示しています。複数のモジュールの連携により、グローバルな情報統合と正確な予測を実現します。まず、入力結晶グラフに対して一連のグラフ畳み込み演算を施し、簡略化されたグラフ構造を生成します。これにより、後続のネットワーク層の計算負荷が軽減され、CGformerの学習プロセスが高速化されます。次に、これに基づいてセンターコードを計算し、結晶グラフのノード特徴を更新します。センターコードには各ノードの入次数と出次数が含まれており、これらは元のノード特徴に統合されます。次に、各ノードはマルチヘッドアテンションモジュール(Multi-head Attention Module)に渡され、可変特徴と空間エンコーディングを組み合わせることで、ノード間の位置関係を表します。センターコードは隣接ノードの平均特徴を和形式に変換し、空間エンコーディングは自己アテンションメカニズムによる隣接ノードの識別を可能にし、効果的なメッセージ集約を促進し、異なる原子間の情報接続を強化します。最後に、出力ベクトルは「プーリング(グローバル特徴の統合)」と「アクティベーション(関数演算)」のプロセスを経て、最終的な材料特性予測が完成します。
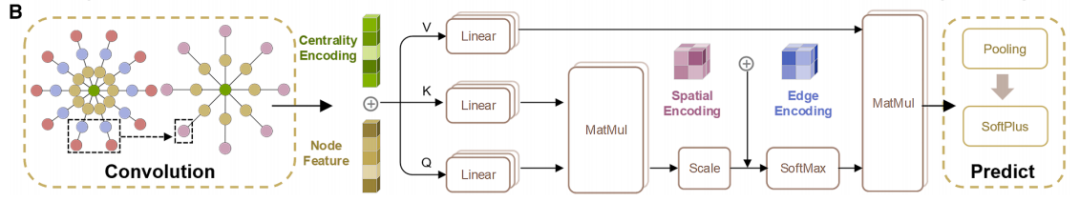
特に注目すべきは、マルチヘッドアテンションモジュールにより、各ノードが結晶グラフ内の隣接ノードだけでなく他のすべてのノードに「注意を払う」ことができるため、長距離の原子間相互作用を捉えることができる点です。さらに、中心エンコーディングと空間エンコーディングの追加により、モデルは原子の化学的性質を特定するだけでなく、構造内における原子の「位置的重要性」と「空間的関係」も認識できるようになり、複雑な結晶の特性評価におけるモデルの精度が向上します。
全体として、従来の結晶ネットワークと比較して、CGformer は質的な飛躍を達成し、グローバルビジョン、情報強化、効率バランスという 3 つの大きな利点を実現し、複雑な高エントロピー材料の発見とパフォーマンスの最適化のための信頼性の高いツールを提供します。
CGformerは強力なパフォーマンスを発揮し、実用的なガイダンスの価値を強調します
CGformerモデルの性能と進歩を正確に評価するため、研究者らはCGCNN、ALIGNN、SchNetといった従来のモデルと比較しました。実験では、「事前学習」と「微調整」の2段階からCGformerの予測精度を検証しました。
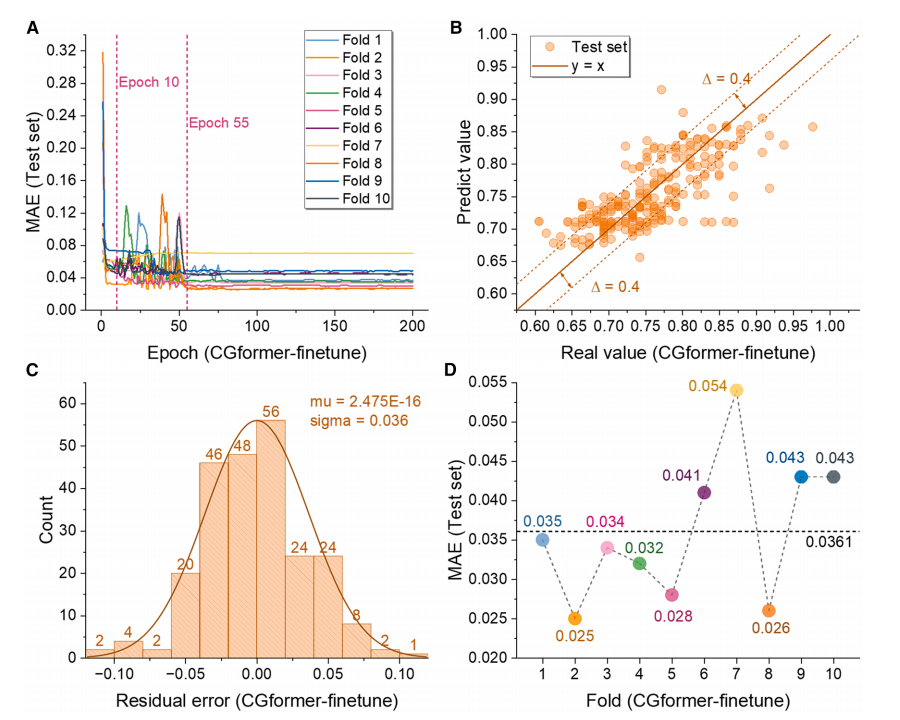
事前学習段階(下図参照)において、CGformerは優れた安定性と予測精度を示しました。初期誤差と変動はCGCNNよりも大幅に低く、10倍交差検証(10倍CV)では、学習セットの平均絶対誤差(MAE)は0.1703で、CGCNNと比較して25.71TP³Tの改善を示しました。また、テストセットの平均MAEは0.3205で、CGCNNと比較して約101TP³Tの改善を示しました。ALIGNNおよびSchNetとの比較においても、CGformerの優れた性能がさらに強調されました。
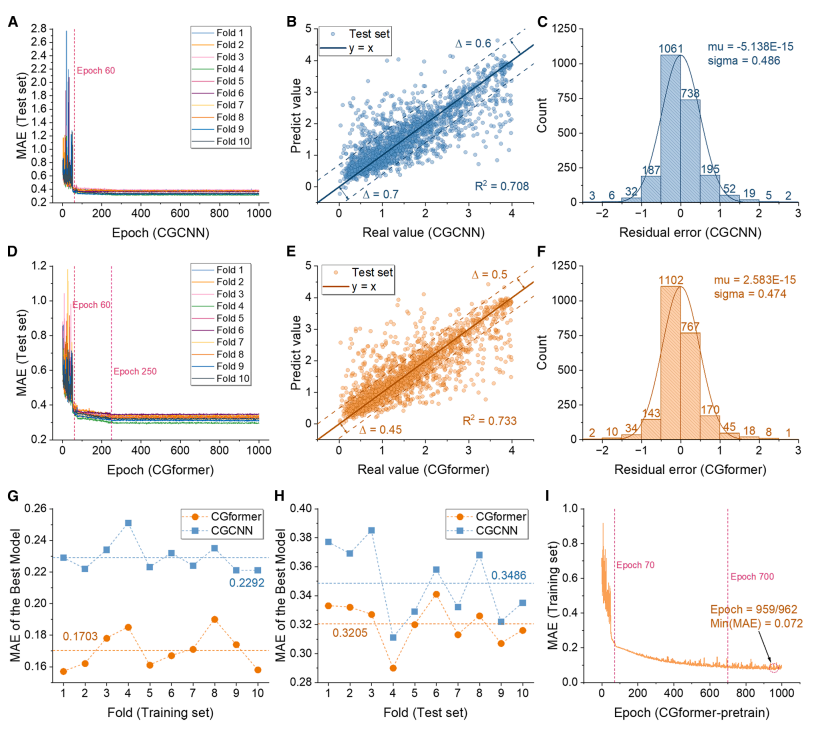
フィッティング結果から、CGformer の予測値は真値からの偏差が少なく、残差は 0 付近に集中し、残差標準偏差は小さくなっており、ナトリウムイオン Eb の予測がより信頼できることが証明されています。
微調整段階(下図参照)では、事前学習済みCGformerのMAEは200回の微調整のうち約10回で大幅に低下し、10分割交差検証の最終平均MAEはわずか0.0361でした。微調整後、予測値と真値の偏差はさらに縮小し、残差は主に-0.05~0.05の範囲に集中し、良好な正規分布を示しました。これは、高エントロピーシステムEbの予測精度が極めて高いことを示し、データ欠損シナリオへの応用可能性を反映しています。
最後に、CGformerによって選定された6つの最適なHE-NSEを合成し、電気化学的特性評価を実施して構造と性能を検証しました。その結果、これらの材料は室温で優れたイオン伝導性を示し、ナトリウムイオン伝導率は25℃で0.093~0.256 mS/cmの範囲にあり、未ドープのNa₃Zr₂Si₂PO₁₂よりも大幅に高いことが示されました。
「人工知能+材料」が材料科学開発の主流となっている
「人工知能+材料」は、現在の材料科学分野における最先端の研究方向となっています。人工知能技術と材料の研究開発、設計、応用を融合することで、両分野の融合がもたらす強力な発展ポテンシャルと応用価値を実証しています。 CGformerの導入は、材料科学分野における人工知能の応用に間違いなく大きな貢献を果たしました。その独自の革新的なアルゴリズムアーキテクチャにより、高エントロピー材料の研究開発における主要な課題を解決します。
CGformerは、AIMS-Labにおける人工知能と材料科学分野の探求における氷山の一角に過ぎません。AIMS-Labの主要な研究分野の一つである人工知能と材料科学の学際的研究は、長年にわたり当研究室の目玉となり、実りある成果を生み出してきました。
昨年、同チームは「Transformerは固体電解質のイオン輸送挙動の進化と導電性の制御を可能にする」と題した研究成果を、主要な国際誌であるEnergy Storage Materialsに発表しました。この研究では、Transformerネットワークアーキテクチャを活用したT-AIMDと呼ばれる人工知能モデルが提案されています。このモデルは、計算コストを大幅に削減すると同時に、あらゆる結晶構造におけるあらゆるイオンの挙動を迅速かつ正確に予測することを可能にします。このアプローチにより、従来のAIMDシミュレーションの速度が100倍以上向上し、材料特性評価プロセスが大幅に加速されます。
用紙のアドレス:
https://www.sciencedirect.com/science/article/abs/pii/S2405829724003829
一方、ドイツのベルリン工科大学とルクセンブルク大学のチームは、AIP Publishingに関連研究を発表し、原子システムのシミュレーションに特化したディープラーニングアーキテクチャ「SchNet」を提案しました。このアーキテクチャは、連続フィルタリング畳み込み層を用いて、周期表における原子種の化学的に妥当な埋め込みを学習し、分子や材料の様々な化学的性質を予測する強力な能力を実証しています。論文のタイトルは「SchNet – 分子と材料のためのディープラーニングアーキテクチャ」です。
用紙のアドレス:
日常的なプラスチック包装や金属製品から、ハイエンド産業におけるナノ材料や超伝導体に至るまで、人類文明の進歩は材料科学の発展と密接に結びついています。人工知能の急速な発展は、材料科学の未来に計り知れない可能性を解き放ち、ひいては人類文明の発展を間接的に推進するでしょう。








