Command Palette
Search for a command to run...
De 100 000 Possibilités À Une Synthèse Réussie, l'intégration Innovante Des Mécanismes d'attention Globale Et Du Modèle d'IA CGformer Facilite Le Développement De Matériaux À Haute entropie.

L'intelligence artificielle transforme profondément le paradigme de la recherche et du développement en science des matériaux, démontrant une valeur révolutionnaire pour accélérer la découverte de nouveaux matériaux et optimiser leurs performances. Grâce à l'intégration étroite du calcul à haut débit et de l'apprentissage automatique, les difficultés des méthodes traditionnelles d'essais-erreurs, telles que les longs cycles expérimentaux et la forte consommation de ressources, ont été efficacement surmontées.L'exploration des matériaux est entrée dans la phase itérative efficace de la « vérification expérimentale pilotée par le calcul ».Cependant, avec l'innovation de la technologie et du mode de vie humains, les exigences de performance des nouveaux matériaux dans des domaines tels que les nouvelles énergies et l'aérospatiale deviennent de plus en plus strictes, et les limites des méthodes traditionnelles d'apprentissage automatique deviennent progressivement apparentes, en particulier dans le domaine de la recherche et du développement de matériaux à haute entropie.
Les matériaux à haute entropie constituent une nouvelle classe de matériaux composés d'un mélange de plusieurs éléments principaux. Grâce à l'action synergique de ces éléments, ils augmentent significativement l'entropie configurationnelle (c'est-à-dire le désordre) de leur arrangement atomique, ce qui leur confère des propriétés mécaniques, une résistance aux hautes températures et une résistance à la corrosion supérieures à celles des matériaux traditionnels. Ces matériaux présentent un potentiel d'application important pour le stockage d'énergie, l'aérospatiale et les équipements pour environnements extrêmes.
Les approches précédentes, telles que les réseaux neuronaux convolutifs à graphes cristallins (CGCNN) et les réseaux neuronaux à graphes linéaires atomistiques (ALIGNN), présentent toutes des défauts architecturaux.Limité par le mécanisme d'interaction des informations locales, il est difficile de modéliser l'effet de synergie atomique à longue distance et ne peut pas capturer entièrement les informations globales propres aux structures cristallines complexes, ce qui entraîne une précision de prédiction limitée.Dans le même temps, les propriétés inhérentes aux matériaux à haute entropie font que leur recherche et leur développement sont confrontés à des défis bien supérieurs à ceux des matériaux traditionnels.Des microstructures complexes, des données expérimentales rares et de haute qualité et un comportement atomique dynamiquement désordonné constituent ensemble des obstacles majeurs au développement de matériaux à haute entropie.
En réponse aux défauts des outils et aux mises à niveau de la demande,L'équipe du professeur Li Jinjin et du professeur Huang Fuqiang du laboratoire d'intelligence artificielle et de microstructure (AIMS-Lab) de l'université Jiao Tong de Shanghai a développé un nouveau modèle de conception de matériaux IA CGformer, qui dépasse avec succès les limites des modèles traditionnels.Ce modèle combine de manière innovante le mécanisme d'attention globale de Graphormer avec CGCNN et intègre le codage de centralité et le codage spatial, de sorte qu'il peut non seulement décrire intuitivement la structure du matériau à travers des diagrammes cristallins, mais également capturer les interactions entre les atomes à longue distance à l'aide du mécanisme « d'attention globale », obtenant ainsi des capacités de traitement de l'information globale que les modèles traditionnels « se concentrant uniquement sur les atomes adjacents » n'ont pas.
Cette méthode fournit des informations structurelles plus complètes, permettant une prédiction plus précise de la migration des ions au sein des structures et offrant un outil fiable pour la recherche et le développement de nouveaux matériaux, notamment les matériaux cristallins complexes et à haute entropie. Les résultats de cette recherche ont été publiés dans la revue de référence Matter sous le titre « CGformer : Réseau de graphes cristallins amélioré par transformateur avec une attention globale pour la prédiction des propriétés des matériaux ».
Points saillants de la recherche :
* La recherche et le développement de CGformer, un modèle de conception de matériaux IA basé sur un mécanisme d'attention globale, fournissent un outil fiable et puissant pour la recherche et le développement des matériaux, contribuant à accélérer le processus de découverte de structures cristallines complexes.
* Comparé au CGCNN, CGformer présente une réduction de 25% de l'erreur absolue moyenne dans l'étude des électrolytes solides à ions sodium à haute entropie (HE-NSE), démontrant ainsi efficacement sa praticité et son avancement.
*18 structures à haute entropie possibles ont été sélectionnées parmi 148 995 et six électrolytes solides à ions sodium à haute entropie (HE-NSE) ont été synthétisés et vérifiés avec succès, avec une conductivité des ions sodium à température ambiante pouvant atteindre 0,256 mS/cm, démontrant ainsi leur valeur d'application pratique.

Adresse du document :
https://www.cell.com/matter/abstract/S2590-2385(25)00423-0
Suivez le compte officiel et répondez « CGformer » pour obtenir le PDF complet
Autres articles sur les frontières de l'IA :
Les ensembles de données multi-catégories améliorent les capacités du modèle CGformer
L’objectif de cette étude est de relever les défis posés par la rareté des données et la complexité structurelle des systèmes à haute entropie grâce à des solutions basées sur des cas.Ce cas de recherche porte sur les véhicules électriques à énergie nouvelle et les applications de stockage d'énergie sur réseau, et plus particulièrement sur la prédiction des performances et le criblage d'électrolytes solides sodium-ion à haute entropie. Différents jeux de données ont été construits et utilisés pour l'apprentissage, le réglage fin et la vérification expérimentale du modèle CGformer, comme suit :
Ensemble de données de base sur la barrière énergétique de diffusion des ions sodium (Eb) :Il s'agit du plus grand ensemble de données connu sur les barrières de diffusion des ions sodium dans les structures à haute entropie, construit par les chercheurs pour cette étude. Il est basé sur les méthodes d'analyse cristallographique par décomposition de Voronoï (CAVD) et d'énergie de site de valence de liaison (BVSE). Cet ensemble de données est principalement utilisé pour le pré-apprentissage de CGformer, permettant au modèle d'apprendre des informations graphiques relatives aux structures contenant du sodium, lesquelles sont ensuite transférées vers l'ensemble de données à haute entropie calculées, jetant ainsi les bases de la prédiction ultérieure de l'Eb des matériaux à haute entropie.
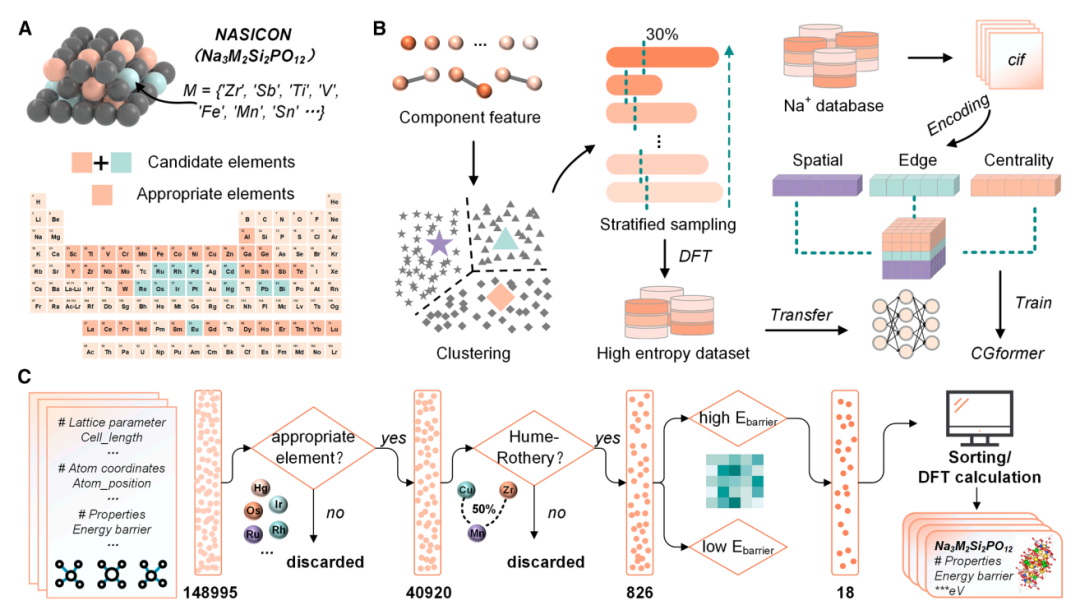
Ensemble de données de calcul HE-NSE :Français Sur la base de Na₃Zr₂Si₂PO₁₂ (comme indiqué ci-dessus), 45 éléments dopants potentiels à haute entropie ont été considérés sur le site Zr, résultant en un espace chimique initial contenant 148 995 structures possibles à haute entropie. Par la suite, grâce à plusieurs cycles de criblage, y compris l'exclusion d'éléments inappropriés (éléments radioactifs, hautement toxiques et coûteux) et des contraintes sur les différences de rayon atomique et l'équilibre de charge, l'espace chimique a été encore réduit à 826 structures relativement stables. Un clustering hiérarchique non supervisé a ensuite été utilisé pour classer ces structures en 20 groupes. De chaque groupe, 301 structures TP3T (238 au total) ont été échantillonnées hiérarchiquement et leurs valeurs Eb ont été calculées en utilisant la théorie de la fonctionnelle de la densité (DFT). Cela a finalement formé un ensemble de données dédié au réglage fin de CGformer, en adaptant spécifiquement le modèle à la tâche de prédiction de l'Eb des ions sodium dans les structures NASICON à haute entropie, améliorant ainsi la précision du modèle dans le scénario cible.
Ensemble de données d'évaluation de la stabilité thermique :Les chercheurs ont extrait de la base de données du Projet Matériaux toutes les structures contenant du sodium dont l'énergie est supérieure à la valeur de l'enveloppe convexe (Ehull) et les ont compilées dans un ensemble d'apprentissage dédié. Cet ensemble de données sert principalement à entraîner un modèle complémentaire d'évaluation de la stabilité thermodynamique des HE-NSE. Associé à l'Eb prédit par CGformer, ce modèle permet de sélectionner des matériaux candidats pour leurs performances et leur stabilité.
L'architecture de fusion innovante permet la « perception globale » de CGformer
CGformer a apporté des innovations fondamentales pour remédier aux lacunes des méthodes traditionnelles, en intégrant deux technologies avancées pour obtenir des avantages complémentaires.Son objectif principal est de conserver la capacité de représentation graphique de la structure cristalline et de briser la limitation consistant à se concentrer uniquement sur les interactions atomiques locales grâce au mécanisme d'attention globale.Plus précisément, il combine le mécanisme d'attention globale de Graphormer avec la méthode de représentation graphique en cristal de CGCNN, tout en ajoutant des modules d'encodage clés pour construire un nouveau pipeline de traitement de l'information.
La figure a ci-dessous montre le processus de codage du diagramme cristallin.Le processus consiste à convertir la structure cristalline tridimensionnelle réelle en un diagramme cristallin qui peut être traité par le modèle.Les atomes d'une structure cristalline sont représentés par des nœuds, et les liaisons chimiques entre eux par des arêtes. Grâce à ce processus de conversion, les chercheurs ont pu extraire des caractéristiques des nœuds et des arêtes, telles que diverses propriétés élémentaires, la charge, le rayon covalent, les distances interatomiques, les types de liaisons et les informations sur la symétrie cristalline. Ces caractéristiques ont ensuite été combinées pour obtenir les données d'entrée initiales requises par CGformer, garantissant ainsi la préservation intégrale des informations chimiques et structurales du cristal.
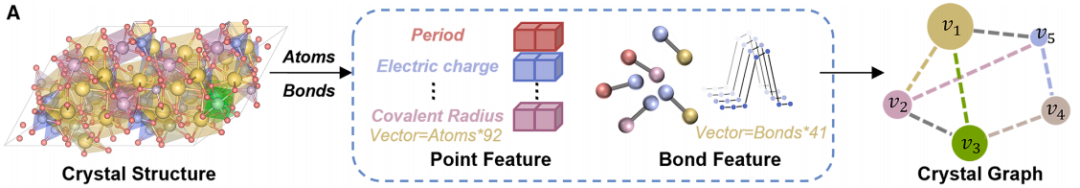
La figure b ci-dessous montre l'architecture réseau de CGformer, grâce à la collaboration multi-modules, l'intégration des informations globales et la prédiction précise sont obtenues.Tout d'abord, le graphe cristallin d'entrée subit une série d'opérations de convolution afin de générer une structure simplifiée, réduisant ainsi la charge de calcul des couches réseau suivantes et accélérant le processus d'apprentissage de CGformer. Ensuite, sur cette base, les chercheurs calculent le code central et mettent à jour les caractéristiques des nœuds du graphe cristallin. Le code central inclut les degrés entrant et sortant de chaque nœud, qui sont ensuite intégrés aux caractéristiques du nœud d'origine. Chaque nœud est ensuite soumis à un module d'attention multi-têtes (Multi-head Attention Module), combinant des caractéristiques variables et un codage spatial pour représenter la relation de position entre les nœuds. Le code central convertit les caractéristiques moyennes des nœuds adjacents en une somme, tandis que le codage spatial permet au mécanisme d'auto-attention de distinguer les nœuds adjacents, de favoriser une agrégation efficace des messages et d'améliorer les connexions d'information entre les différents atomes. Enfin, le vecteur de sortie subit les processus de « pooling (intégration des caractéristiques globales) » et d'« activation (opération de fonction) » pour finaliser la prédiction finale des propriétés du matériau.
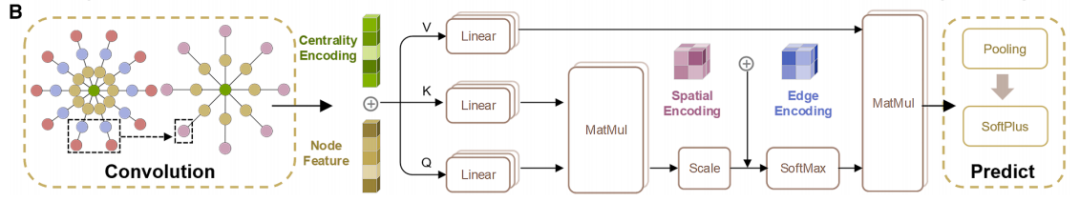
Notamment, le module d'attention multi-têtes permet à chaque nœud de prêter attention à tous les autres nœuds du graphe cristallin, plutôt qu'aux nœuds adjacents uniquement, capturant ainsi les interactions atomiques à longue portée. De plus, l'ajout du codage central et du codage spatial permet au modèle non seulement d'identifier les propriétés chimiques des atomes, mais aussi de percevoir leur importance positionnelle et leurs relations spatiales au sein de la structure, améliorant ainsi la précision du modèle dans la caractérisation des cristaux complexes.
en tout,Par rapport aux réseaux cristallins traditionnels, CGformer a réalisé un saut qualitatif, réalisant trois avantages majeurs : une vision globale, une amélioration de l'information et un équilibre d'efficacité, fournissant un outil crédible et fiable pour la découverte et l'optimisation des performances de matériaux complexes à haute entropie.
CGformer démontre des performances puissantes et met en évidence sa valeur d'orientation pratique
Afin d'évaluer avec précision les performances et l'évolution du modèle CGformer, les chercheurs l'ont comparé à des modèles traditionnels tels que CGCNN, ALIGNN et SchNet. L'expérience a permis de vérifier la précision des prédictions de CGformer en deux étapes : le « pré-apprentissage » et le « réglage fin ».
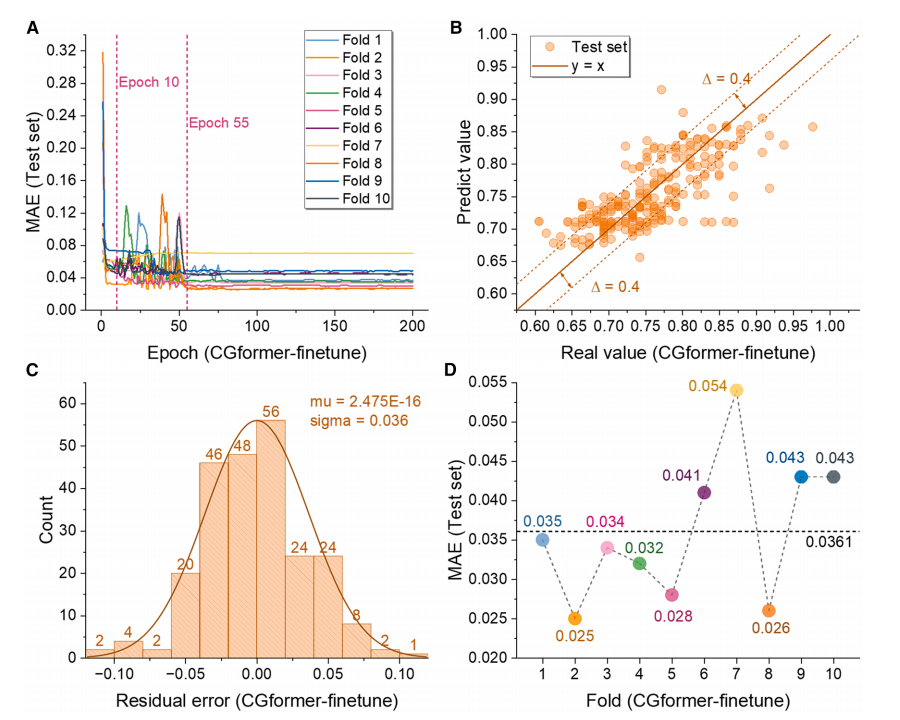
Durant la phase de pré-apprentissage (comme illustré dans la figure ci-dessous), CGformer a démontré une stabilité et une précision de prédiction supérieures. Son erreur initiale et sa fluctuation étaient significativement inférieures à celles de CGCNN. Une validation croisée décuplée (CV décuplé) a montré que l'erreur absolue moyenne (EMA) de son ensemble d'apprentissage était de 0,1703, soit une amélioration de 25,71 TP³T par rapport à CGCNN ; l'EMA moyenne de son ensemble de test était de 0,3205, soit une amélioration de près de 101 TP³T par rapport à CGCNN. Les comparaisons avec ALIGNN et SchNet ont également mis en évidence les performances supérieures de CGformer.
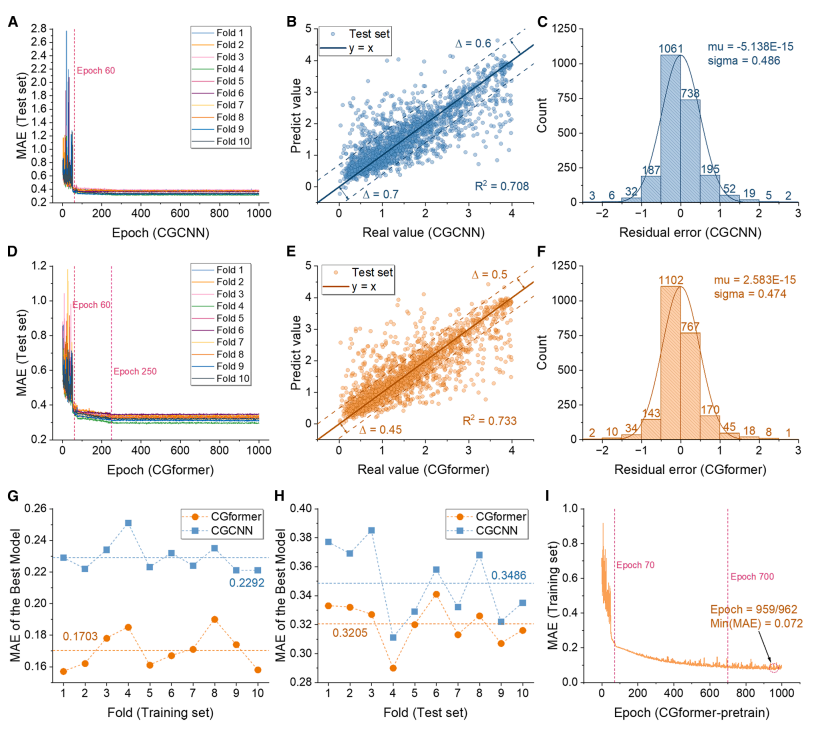
D'après les résultats d'ajustement, la valeur prédite de CGformer s'écarte moins de la valeur réelle, le résidu est plus concentré près de 0 et l'écart type résiduel est plus petit, ce qui prouve que sa prédiction de l'ion sodium Eb est plus fiable.
Durant la phase de réglage fin (comme illustré dans la figure ci-dessous), l'écart-type moyen (MAE) du CGformer pré-entraîné a diminué significativement après environ 10 cycles de réglage fin sur 200, et l'écart-type moyen final de la validation croisée décuplée n'était que de 0,0361. Après le réglage fin, l'écart entre la valeur prédite et la valeur réelle a encore diminué, et les résidus se sont principalement concentrés dans la plage de -0,05 à 0,05, présentant une bonne distribution normale, démontrant sa très grande précision dans la prédiction du système à haute entropie Eb et reflétant son potentiel d'application dans les scénarios où les données sont manquantes.
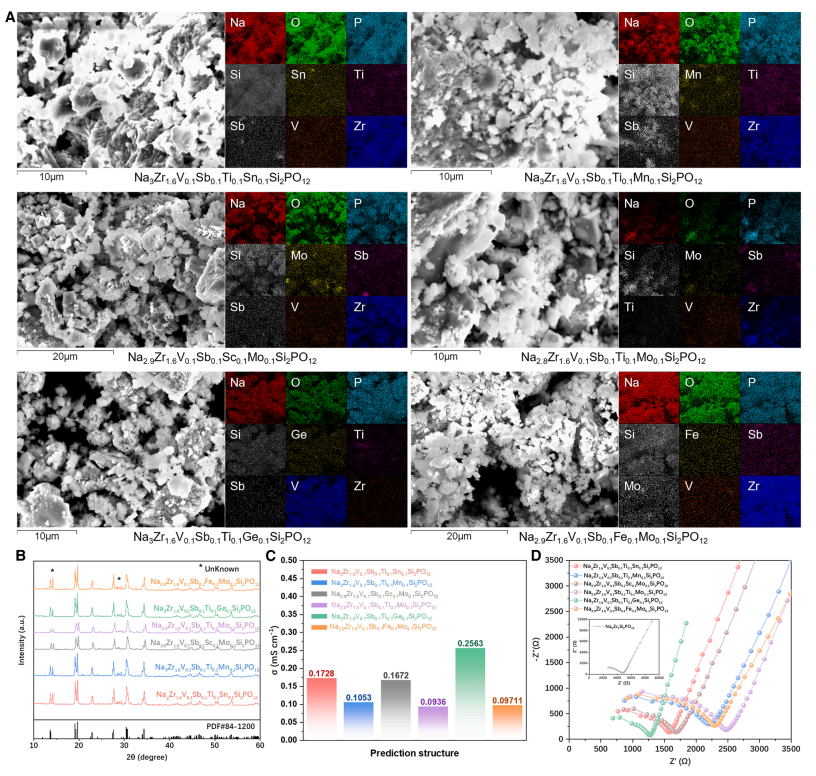
Enfin, les six HE-NSE optimaux sélectionnés par CGformer ont été synthétisés et caractérisés électrochimiquement afin de vérifier leur structure et leurs performances. Les résultats ont montré que ces matériaux présentaient une excellente conductivité ionique à température ambiante, la conductivité des ions sodium variant de 0,093 à 0,256 mS/cm à 25 °C, significativement supérieure à celle du Na₃Zr₂Si₂PO₁₂ non dopé.
« Intelligence artificielle + matériaux » est devenu le courant dominant du développement de la science des matériaux
L'alliance « intelligence artificielle et matériaux » est devenue une orientation de recherche de pointe dans le domaine actuel des sciences des matériaux. L'intégration de l'intelligence artificielle à la recherche et au développement, à la conception et à l'application des matériaux démontre le fort potentiel de développement et la valeur applicative de l'intersection de ces deux disciplines. L'introduction de CGformer a incontestablement contribué de manière significative à l'application de l'intelligence artificielle dans le domaine des sciences des matériaux. Grâce à son architecture algorithmique unique et innovante, il résout les problèmes clés de la recherche et du développement de matériaux à haute entropie.
CGformer n'est que la partie émergée de l'iceberg des recherches menées par AIMS-Lab dans les domaines de l'intelligence artificielle et de la science des matériaux. L'un des axes de recherche majeurs du laboratoire, la recherche interdisciplinaire en intelligence artificielle et en science des matériaux est depuis longtemps devenue une référence incontournable et a produit des résultats fructueux.
L'année dernière, la même équipe a présenté ses résultats de recherche dans la revue internationale de référence Energy Storage Materials, sous le titre « Transformer permet l'évolution du comportement du transport ionique et la régulation de la conductivité des électrolytes solides ». L'étude proposait un modèle d'intelligence artificielle appelé T-AIMD, qui utilise une architecture réseau Transformer. Ce modèle réduit considérablement les coûts de calcul tout en permettant une prédiction rapide et précise du comportement de tout ion dans toute structure cristalline. Cette approche multiplie par plus de 100 la vitesse des simulations AIMD traditionnelles, accélérant ainsi considérablement le processus d'évaluation des propriétés des matériaux.
Adresse du document :
https://www.sciencedirect.com/science/article/abs/pii/S2405829724003829
Par ailleurs, des équipes de l'Université technique de Berlin et de l'Université du Luxembourg (Allemagne) ont publié des recherches connexes dans AIP Publishing, proposant SchNet, une architecture d'apprentissage profond spécialement conçue pour la simulation de systèmes atomiques. Cette architecture utilise des couches convolutives à filtrage continu pour apprendre des inclusions chimiquement plausibles de types atomiques dans le tableau périodique, démontrant ainsi de puissantes capacités de prédiction de diverses propriétés chimiques des molécules et des matériaux. L'article est intitulé « SchNet – Une architecture d'apprentissage profond pour les molécules et les matériaux ».
Adresse du document :
Des emballages plastiques et produits métalliques courants aux nanomatériaux et supraconducteurs utilisés dans les industries de pointe, le progrès de l'humanité est étroitement lié au développement de la science des matériaux. Le développement rapide de l'intelligence artificielle ouvrira sans aucun doute un potentiel considérable à l'avenir de la science des matériaux, ce qui, indirectement, stimulera le progrès de l'humanité.








