Command Palette
Search for a command to run...
Sélectionné Pour l'ICLR 2025 ! Shen Chunhua Et d'autres De l'Université Du Zhejiang Ont Proposé La Technologie d'alignement De Boltzmann, Et La Prédiction De l'énergie Libre De Liaison Aux Protéines a Atteint SOTA

Les interactions protéine-protéine (IPP) sont la base sur laquelle tous les organismes peuvent exécuter diverses fonctions biologiques, qui sont principalement réalisées grâce à l'interaction et à l'influence entre différentes molécules de protéines. L’identification et la compréhension précises des interactions protéine-protéine sont extrêmement importantes pour déchiffrer les fonctions des protéines, révéler les activités de la vie, explorer les mécanismes des maladies, développer des médicaments ciblés et innover dans les applications biologiques.
Avec le développement des ordinateurs et de l’intelligence artificielle, la recherche sur les IPP dans la communauté de recherche scientifique a fait de grands progrès ces dernières années avec le soutien de l’apprentissage profond. En particulier AlphaFold 3 publié par DeepMind en 2024,Le taux de réussite de la prédiction de la structure des complexes protéiques généraux a été porté à près de 80%.Cela résout également efficacement le problème de la modélisation informatique haute fidélité des interactions protéiques qui a tourmenté la communauté de recherche scientifique pendant des décennies.
Cependant, l’interaction entre les protéines est un processus dynamique qui comprend la liaison et la dissociation. Il est difficile de saisir pleinement l’interaction entre les molécules biologiques en étudiant uniquement les structures statiques.Des paramètres tels que l'énergie libre de liaison (∆G, la différence d'énergie libre de Gibbs entre les états liés et non liés) peuvent caractériser quantitativement la dynamique des interactions protéine-protéine.Cependant, la manière de prédire avec précision le changement de l'énergie libre de liaison (∆∆G, également connu sous le nom d'effet de mutation) est devenue l'une des conditions préalables pour que la communauté scientifique comprenne ou régule les interactions protéine-protéine.
Sur cette base, l'équipe du professeur Shen Chunhua de l'École d'informatique et de technologie de l'Université du Zhejiang, en collaboration avec des équipes de l'Université d'Adélaïde en Australie et de l'Université Northeastern aux États-Unis,Nous proposons conjointement une technique appelée alignement de Boltzmann pour transférer les connaissances d'un modèle de pliage inverse pré-entraîné aux prédictions de ∆∆G.L'étude a d'abord analysé la définition thermodynamique de ∆∆G et a lié la distribution de conformation de l'énergie et des protéines en introduisant la distribution de Boltzmann, mettant ainsi en évidence le potentiel des modèles probabilistes pré-entraînés. L'équipe a ensuite utilisé le théorème de Bayes pour contourner l'estimation directe et a utilisé la vraisemblance logarithmique fournie par le modèle de repliement inverse des protéines pour estimer ∆∆G. Cette dérivation fournit une explication rationnelle de la forte corrélation entre l’énergie de liaison et la vraisemblance logarithmique du modèle de repliement inverse observé dans d’autres expériences précédentes.
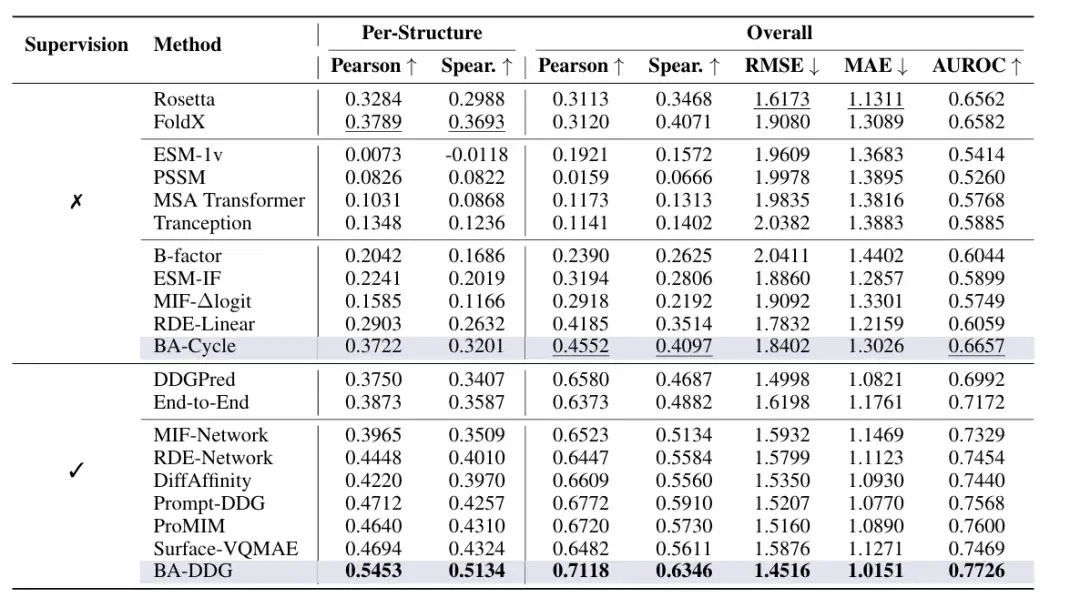
Par rapport à la méthode précédente basée sur le pliage inverse, les résultats expérimentaux de cette méthode sur l'ensemble de données SKEMPI v2 montrent un niveau supérieur.Son coefficient de Spearman dans les états supervisés et non supervisés a atteint respectivement 0,5134 et 0,3201.Significativement plus élevé que les méthodes SOTA précédentes de 0,4324 et 0,2632.
Cette réalisation, intitulée « Boltzmann-Aligned Inverse Folding Model as a Predictor of Mutational Effects on Protein-Protein Interactions », a été incluse dans l'ICLR 2025, la principale conférence universitaire internationale dans le domaine de l'intelligence artificielle. Il convient de mentionner que l'ICLR de cette année a reçu un total de 11 565 soumissions, et que seulement 32 081 manuscrits ont été acceptés.

Adresse du document :
https://arxiv.org/abs/2410.09543
Recommander un événement de partage académique. La dernière invitation à la diffusion en direct de Meet AI4S aura lieu à 12h00 le 7 mars.Huang Hong, professeur associé à l'Université des sciences et technologies de Huazhong, Zhou Dongzhan, jeune chercheur au Centre d'IA pour les sciences du Laboratoire d'intelligence artificielle de Shanghai, et Zhou Bingxin, chercheur adjoint à l'Institut des sciences naturelles de l'Université Jiao Tong de Shanghai,Présentez vos réalisations personnelles et partagez votre expérience de recherche scientifique.
L'apprentissage profond accélère le changement de paradigme dans le calcul des effets des mutations
La communauté scientifique étudie depuis longtemps la prédiction de ∆∆G.Les méthodes traditionnelles peuvent être divisées en deux catégories : les méthodes biophysiques et les méthodes statistiques.Parmi elles, les méthodes biophysiques simulent principalement la manière dont les protéines interagissent au niveau atomique grâce à des calculs énergétiques ; Les méthodes statistiques s'appuient sur l'ingénierie des caractéristiques, utilisant principalement des descripteurs pour capturer les caractéristiques géométriques, physiques et évolutives des protéines.
Il ne fait aucun doute que quelle que soit la méthode traditionnelle utilisée, elle doit s’appuyer fortement sur l’expertise humaine, ce qui est non seulement chronophage et exigeant en main-d’œuvre, mais également incapable de capturer avec précision les interactions complexes entre les protéines. De plus, les deux méthodes ont leurs propres inconvénients. Par exemple, les méthodes biophysiques sont souvent confrontées à des défis pour équilibrer la vitesse et la précision. Les méthodes basées sur l’apprentissage profond montrent non seulement un grand « talent » dans la modélisation des protéines, mais accélèrent également la transformation du paradigme de prédiction ∆∆G.
De plus en plus de cas le prouvent. Par exemple, une équipe de l’Académie chinoise des sciences a proposé une méthode basée sur l’apprentissage de la représentation appelée SidechainDiff.Cette méthode utilise le modèle de diffusion de Riemann pour apprendre le processus de génération des conformations de la chaîne latérale et peut également donner une représentation structurelle de fond des mutations à l'interface protéine-protéine.En utilisant les représentations apprises, la méthode atteint des performances de pointe dans la prédiction des effets des mutations sur la liaison protéine-protéine.
Ce résultat est intitulé « Prédire les effets mutationnels sur la liaison protéine-protéine via un modèle probabiliste de diffusion de chaîne latérale » et est inclus dans NeurIPS 2023.
* Adresse du papier :
Bien que les méthodes basées sur l’apprentissage profond aient obtenu des résultats considérables, elles ne sont pas parfaites. Par coïncidence avec l’exemple ci-dessus,Cet article mentionne également qu'il existe un manque de données expérimentales pour expliquer l'énergie de liaison.Ceci est généralement considéré comme un défi majeur basé sur les méthodes d’apprentissage en profondeur, ce qui a directement conduit davantage d’équipes à se pré-entraîner sur un grand nombre d’ensembles de données non étiquetés avant d’améliorer la capacité à prédire les mutations. Cela implique une variété de tâches d'agent de pré-formation, telles que le repliement inverse des protéines, la modélisation de masques et la modélisation de chaînes latérales dans l'exemple ci-dessus.
Heureusement, ces méthodes « alternatives » ont atteint leurs objectifs, mais malheureusement, elles ont également montré leurs faiblesses sans exception. La plupart des méthodes basées sur la pré-formation utilisent uniquement le réglage fin supervisé (SFT).Cependant, l’importance de l’alignement des données est ignorée, ce qui peut amener le réglage fin supervisé à faire oublier au modèle les connaissances générales précédemment acquises lors de la pré-formation non supervisée, ce qui entraîne un risque de surajustement.Avec le recul, ces méthodes « alternatives » soulignent sans aucun doute l’urgence de transférer les connaissances acquises pour une prédiction précise des mutations.
Développement innovant de l'alignement de Boltzmann pour surpasser les modèles SOTA
Plus précisément, l’équipe de recherche s’est d’abord basée sur les principes de la distribution de Boltzmann et du cycle thermodynamique,Le changement de l'énergie libre de liaison lorsqu'une protéine mute est lié à la probabilité d'apparition de la séquence d'acides aminés de la protéine.L'alignement de Boltzmann a été proposé (comme indiqué sur le côté droit de la figure ci-dessous). Par la suite, l'équipe de recherche a proposé une méthode appelée BA-Cycle, qui intégrait le modèle de pliage inverse dans l'alignement de Boltzmann et utilisait le modèle de pliage inverse pour évaluer les mutations en prédisant la probabilité des séquences protéiques (comme indiqué sur le côté gauche de la figure ci-dessous).
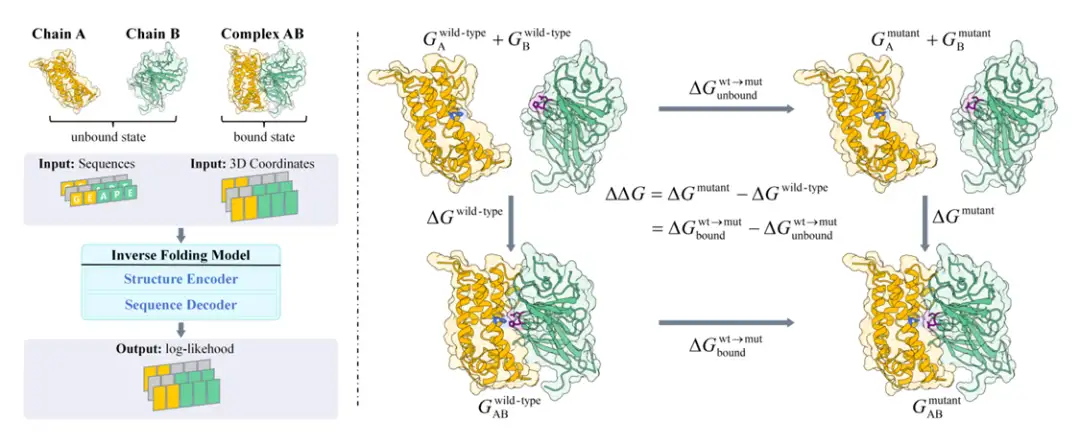
Il convient de mentionner que pour établir le lien entre l'énergie libre de liaison aux protéines et la probabilité conditionnelle de la séquence protéique, et pour résoudre les deux principales difficultés rencontrées lors de l'estimation directe de la probabilité p(X|S) de la structure des protéines sous une séquence donnée :Les limites des modèles existants de prédiction de la structure des protéines et les lacunes des modèles probabilistes,L'équipe de recherche a remplacé le théorème de Bayes dans la formule de calcul de la liberté de liaison, c'est-à-dire p(X|S) = p(S|X) ・ p(X)/p(S), et a réussi à lier l'énergie libre de liaison à la probabilité conditionnelle p (X|S) de la séquence protéique, évitant ainsi la difficulté d'estimer directement p (X|S). Cela a jeté les bases d’une analyse plus approfondie de la relation entre les changements dans l’énergie libre de liaison et la probabilité conditionnelle des séquences protéiques.
De plus, comme on suppose que la structure de la protéine reste inchangée avant et après la mutation,L’équipe de recherche a utilisé le modèle de pliage inverse pour évaluer les probabilités de séquence des états liés et non liés.La structure de base de l’état lié est généralement connue et le modèle peut calculer directement sa probabilité ; la structure de l'épine dorsale de l'état non lié n'est pas explicitement donnée, et la probabilité peut être estimée en évaluant les deux chaînes du complexe séparément.
Sur cette base,L'équipe de recherche a proposé une méthode appelée BA-Cycle pour l'estimation non supervisée de ∆∆G.L'évaluation non supervisée de ∆∆G a été réalisée à l'aide du modèle de pliage inverse pré-entraîné ProteinMPNN. Cela contraste fortement avec les études précédentes sur le même sujet qui ne prenaient pas explicitement en compte la probabilité d’états non liés dans le cycle thermodynamique.
enfin,L’équipe de recherche a également proposé une méthode appelée BA-DDG.Le cycle BA a été affiné par l'alignement de Boltzmann en utilisant les données de changement d'énergie libre de liaison. BA-DDG utilise le même processus direct que BA-Cycle. L’objectif de BA-DDG est de minimiser l’écart entre le véritable changement d’énergie libre de liaison et le changement d’énergie libre de liaison prévu tout en maintenant la distribution du modèle pré-entraîné d’origine.
L’équipe de recherche a mené une série de vérifications expérimentales sur l’ensemble de données SKEMPI v2.Parmi eux, l'ensemble de données SKEMPI v2 est un ensemble de données de mutation annotées contenant 348 complexes protéiques, dont 7 085 mutations d'acides aminés ainsi que des changements dans les paramètres thermodynamiques et les constantes de vitesse cinétique.
Il existe au total 7 indicateurs d'évaluation, dont 5 indicateurs globaux, à savoir le coefficient de corrélation de Pearson, le coefficient de corrélation de rang de Spearman, l'erreur quadratique moyenne minimale (RMSE), l'erreur absolue moyenne minimale (MAE) et l'AUROC. De plus, l’équipe de recherche a regroupé les mutations en fonction de leurs caractéristiques structurelles et a calculé le coefficient de corrélation de Pearson et le coefficient de corrélation de Spearman pour chaque groupe comme deux indicateurs supplémentaires.
L'équipe de recherche a d'abord comparé BA-Cyale et BA-DDG avec les méthodes SOTA non supervisées et supervisées,Il existe trois types de méthodes non supervisées, notamment les fonctions énergétiques empiriques traditionnelles telles que Rosetta Cartésian ∆∆G et FoldX ; méthodes basées sur la séquence/l'évolution telles que ESM-1v, Position-Specific Scoring Matrix (PSSM), MSA Transformer et Tranception ; et des méthodes pré-entraînées basées sur des informations structurelles qui ne sont pas formées sur les étiquettes ∆∆G, telles que ESM-1F, MIF-∆logits, RDE-Linear et B-factor.
Les méthodes supervisées sont divisées en deux catégories, dont les modèles d’apprentissage de bout en bout tels que DDGPred et End-to-End ; et des méthodes de pré-formation basées sur des informations structurelles, affinées sur ∆∆G, notamment MIF-Network, RDE-Network, DiffAffinity, Prompt-DDG, ProMIM et Surface-VQMAE.
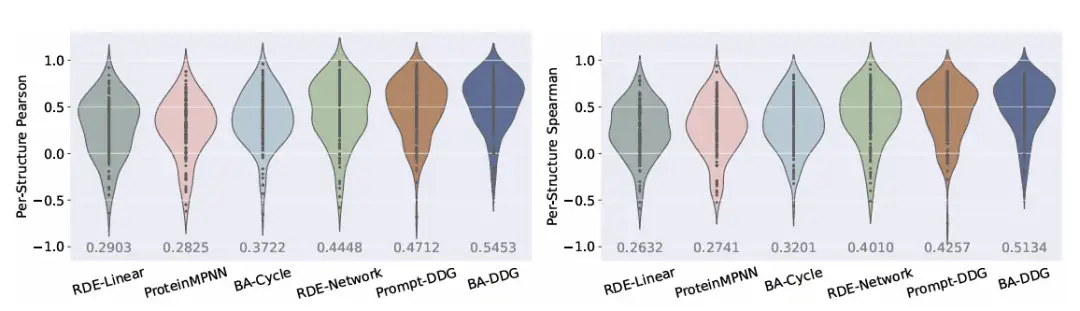
Les résultats montrent queBA-DDG surpasse toutes les valeurs de référence dans toutes les mesures d’évaluation.Parmi eux, le coefficient de corrélation de Pearson et le coefficient de corrélation de Spearman sous la méthode supervisée ont atteint respectivement 0,5453 et 0,5134. Son amélioration significative dans la corrélation de chaque structure met en évidence sa plus grande fiabilité dans les applications pratiques ;BA-Cycle atteint des performances comparables à la fonction énergétique empirique et surpasse toutes les lignes de base d'apprentissage non supervisé.Comme le montre la figure suivante :
En outre, il ressort clairement de l’analyse visuelle pertinente queBA-DDG surpasse les autres méthodes en termes de visualisation qualitative et de mesures quantitatives.Comme le montre la figure suivante :
En outre, les chercheurs ont mené des expériences sur la prédiction de l’énergie de liaison, l’amarrage protéine-protéine et l’optimisation des anticorps, et les résultats ont montré sa large applicabilité. Ces impacts positifs joueront un rôle extrêmement important dans la conception de médicaments et le criblage virtuel, posant les bases théoriques de leur application réelle à l’avenir.
Cultiver en profondeur l'apprentissage automatique et la vision artificielle pour réaliser l'universalisation de l'IA
Dans cette étude, les chercheurs ont utilisé des théories interdisciplinaires pour fournir de nouvelles perspectives pour l’analyse des séquences protéiques et ont en même temps formé un cadre de recherche systématique grâce à l’intégration et à l’optimisation innovantes des modèles. Cette méthode de recherche étape par étape permet non seulement de comprendre pleinement et en profondeur la relation entre la séquence des protéines et les changements d’énergie libre, mais fournit également une nouvelle idée pour les recherches ultérieures.
Il convient de mentionner queLe professeur Shen Chunhua, l’un des principaux participants à cette recherche, s’est engagé depuis longtemps dans la recherche sur l’apprentissage automatique et la vision par ordinateur.Il a publié plus de 150 articles à ce jour, dont certains sur des plateformes universitaires de renommée internationale telles que TPAMI et IJCV. À peine deux mois après le début de l’année 2025, l’équipe dirigée par le professeur Shen Chunhua a produit des résultats importants, en publiant trois articles sur la plateforme de pré-impression arXiv.
Dans le premier article, le groupe de recherche du professeur Shen Chunhua a développé un modèle basé sur l'ADN basé sur le réseau CNN, nommé ConvNova. Le modèle est de conception simple mais offre des performances remarquables.Dans la tâche d'histone associée, le score moyen a dépassé la méthode de deuxième place 5.8%, permettant des calculs plus rapides avec moins de paramètres.Dans le même temps, cette méthode vérifie également que la méthode basée sur l’architecture du réseau CNN présente un fort potentiel concurrentiel par rapport aux méthodes basées sur le réseau Transformer et le réseau SSM. La recherche connexe a été publiée sous le titre « Revisiting Convolution Architecture in the Realm of DNA Foundation Models ».
* Adresse du papier :
https://arxiv.org/abs/2502.18538
Dans le deuxième article, le groupe de recherche du professeur Shen Chunhua et le laboratoire d’IA de Shanghai ont développé conjointement un modèle de vision générale DICEPTION.Le modèle de diffusion pré-entraîné est utilisé pour résoudre les problèmes de perception visuelle multitâches, ce qui nécessite moins de données d'entraînement et présente une forte adaptabilité aux tâches.En utilisant seulement 0,06% de données SAM, le modèle atteint un niveau comparable à celui des modèles SOTA dans des tâches telles que la segmentation, et réduit considérablement les coûts de formation en unifiant les résultats des tâches grâce au codage couleur. La recherche connexe a été publiée sous le titre « DICEPTION : un modèle de diffusion généraliste pour les tâches de perception visuelle ».
* Adresse du papier :
https://arxiv.org/pdf/2502.17157
Dans le troisième article, l’équipe du professeur Shen Chunhua, en collaboration avec Alibaba, a proposé un benchmark appelé PhyCoBench, qui est utilisé pour évaluer la capacité des modèles de génération vidéo à générer des vidéos conformes aux lois de la physique. L'étude présente également le modèle d'évaluation automatique PhyCoPredictor, un modèle de diffusion qui génère un flux optique et des images vidéo en cascade. En comparant l’évaluation de la cohérence du tri automatique et manuel,Les résultats expérimentaux montrent que PhyCoPredictor possède la capacité la plus proche de l’évaluation humaine.La recherche connexe a été publiée sous le titre « Un benchmark de cohérence physique pour l’évaluation des modèles de génération vidéo via la prédiction d’images guidée par flux optique ».
* Adresse du papier :
https://arxiv.org/pdf/2502.05503
L’équipe du professeur Shen Chunhua a non seulement obtenu des résultats fructueux, mais son influence personnelle est également remarquable. Les articles pertinents publiés par le professeur Shen Chunhua ont toujours été une source importante de citations dans la communauté de recherche scientifique. Il a également été sélectionné dans la liste des « 2023 chercheurs chinois les plus cités » publiée par Elsevier, une société mondiale d'analyse d'informations.
Aujourd'hui, le professeur Shen Chunhua occupe depuis trois ans la chaire Qiushi et est directeur adjoint du laboratoire national clé de conception assistée par ordinateur et de systèmes d'image à l'université du Zhejiang. Il a non seulement obtenu des résultats de recherche fructueux, mais aussi des résultats d'enseignement considérables et a formé de nombreux étudiants de master et de doctorat. En outre, le Laboratoire national clé de conception assistée par ordinateur et de systèmes graphiques, où il est situé, sert d'interface reliant « industrie-université-recherche » et a également connu un développement multiforme ces dernières années. Elle a coopéré avec de nombreuses entreprises, dont Ant, et est devenue une base d'innovation pour la recherche scientifique, une base de formation des talents et une base d'incubation d'innovation.








