Command Palette
Search for a command to run...
LAMMPS模拟软件包的一套教程
LAMMPS模拟软件包的一套教程
Simon Gravelle Cecilia M. S. Alvaras Jacob R. Gissinger Axel Kohlmeyer
一键部署 LAMMPS 入门教程:npt 控温估计 FCC Cu 熔点
摘要
开源分子模拟软件包的可用性使科学家和工程师能够专注于运行和分析模拟,而无需编写、并行化和验证自己的模拟软件。虽然这使得分子模拟对更广泛的受众变得可及,但此类软件包的“黑箱”特性以及众多选项和功能可能使其难以正确使用,尤其是对于模拟领域的新手而言。LAMMPS 是这样一个多功能的分子模拟代码,旨在跨广泛的材料科学和计算化学应用领域对基于粒子的系统进行建模,包括原子尺度、粗粒化、介观、无网格连续体和离散元模型。LAMMPS 能够从小型台式计算机到大规模超级计算环境高效运行不同规模的模拟。其灵活性和可扩展性使其成为原子和分子系统乃至更广泛复杂且大规模模拟的理想选择。本文介绍了一套旨在帮助新用户更易学习 LAMMPS 的教程套件。前四个教程涵盖在 LAMMPS 中运行具有不同复杂度系统的分子模拟的基础知识。后四个教程涉及更高级的分子模拟技术,具体包括反应力场的使用、巨正则蒙特卡洛方法、增强采样以及 REACTER 协议。此外,我们介绍了 LAMMPS-GUI,这是一种增强的跨平台图形文本编辑器,专为与 LAMMPS 配合使用而设计,能够直接在编辑后的输入文件上运行 LAMMPS。在本套教程中,LAMMPS-GUI 被用作主要工具,用于编辑输入文件、运行 LAMMPS、提取数据以及可视化模拟系统。
一句话总结
本文介绍了一套结构化的教程套件以及 LAMMPS-GUI,这是一款跨平台的图形化文本编辑器,整合了输入编辑、模拟执行、数据提取和可视化功能。该工具引导用户从基础设置逐步掌握高级技术,如反应力场、巨正则蒙特卡洛模拟、增强采样以及 REACTER 协议,从而提升材料科学与计算化学中基于粒子的建模技术的可及性。
核心贡献
- 本文介绍了一套包含八个教程的套件,引导用户从基础模拟设置过渡到高级技术,包括反应力场、巨正则蒙特卡洛模拟、增强采样以及 REACTER 协议。这些逐步推进的工作流程为 LAMMPS 中复杂的物理模型选择与参数配置提供了标准化操作指南。
- 该研究推出了 LAMMPS-GUI,这是一款跨平台图形化文本编辑器,专为 LAMMPS 工作流整合了输入编辑、直接执行、数据提取与系统可视化功能。该界面作为所有教程中的核心工具,用于管理模拟输入、执行过程及数据分析。
- 具体的实现示例展示了利用自由采样和伞形采样方法计算自由能分布的过程。这些文档化的操作流程为更广泛的应用提供了可适配的模板,涵盖吸附势垒分析与膜转运研究等领域。
引言
分子模拟是材料科学与计算化学的基石,使研究人员能够预测结构、热力学与动力学属性,从而在计算模型与实验数据之间建立桥梁。尽管 LAMMPS 等开源平台提供了模拟复杂原子与粗粒化系统所需的并行计算能力,但其陡峭的学习曲线与密集的文档往往令初学者望而却步。处理复杂的输入脚本、选择合适的力场以及配置热力学系综常常导致进度停滞,实际上形成了一道限制其更广泛科学应用的壁垒。为突破这些瓶颈,作者推出了一套包含八个教程的结构化指南,引导用户从基础模拟逐步进阶至反应力场与增强采样等高级技术。同时发布的还有 LAMMPS-GUI,这是一款跨平台图形编辑器,可简化输入创建、执行与数据可视化流程。这套教育与软件相结合的整体框架有效降低了入门门槛,赋能研究人员可靠地设计与解读分子动力学及蒙特卡洛工作流。
数据集
Dataset Composition and Sources
- 作者提供了一套包含八个教学教程的集合,托管于 GitHub 并通过 LAMMPS-GUI 提供访问入口。
- 运行这些资源需要 LAMMPS 稳定版 v22Jul2025 与 LAMMPS-GUI v1.7.0,支持 Linux、macOS 与 Windows 系统。
- 可视化与分析环节引用了 VMD、OVITO、Python 库及绘图软件等外部工具。
Key Details for Each Subset
- Tutorial 1: 二元 Lennard-Jones 流体系统,包含 1500 个 1 型原子与 100 个 2 型原子。模拟过程执行能量最小化,并在 NVE 与 NVT 系综下运行。
- Tutorial 2: 碳纳米管模型,采用 OPLS-AA 与 AIREBO 力场。系统经历外部形变以演示键断裂过程及可视化效果。
- Tutorial 3: 溶剂化聚合物系统,结合柔性 SPC/Fw 水分子与聚合物链。系统配置包含 700 个水分子,并使用长程静电求解器进行 NPT 系综平衡。
- Tutorial 4: 壁面间的受限电解质,采用刚性 TIP4P/2005 水模型。本教程演示了带有剪切流场的非平衡分子动力学模拟。
- Tutorial 5: 反应系统,采用 ReaxFF 力场以模拟动态化学反应与电荷平衡。
- Tutorial 6: 通过巨正则蒙特卡洛模拟对二氧化硅孔道内的流体吸附进行建模,以模拟与粒子库交换粒子的开放系统。
- Tutorial 7: 高级自由能计算方法,利用伞形采样技术计算难以采样的势能面中的能量势垒。
- Tutorial 8: 尼龙-6,6 聚合模拟,其中碳纳米管嵌入熔体中。REACTER 协议用于追踪随时间推移的聚合过程与水分生成情况。
Data Usage and Processing
- 作者将该集合作为学习资源使用,而非机器学习模型的训练数据集。
- 用户按顺序跟随教程生成模拟输入文件并分析结果。本流程不涉及训练集划分、混合比例或裁剪策略。
- 处理流程包括定义区域、创建具有随机放置与重叠约束的原子,以及通过数据文件导入拓扑结构。
- 元数据在输入文件与数据文件中构建,明确指定原子类型、力场参数及键定义。
- 轨迹可视化通过 dump 命令生成图像序列实现,并提供调整渲染风格与相机视角的选项。
方法
作者利用 LAMMPS-GUI(一款专为创建与执行 LAMMPS 输入脚本设计的图形化文本编辑器)来简化分子动力学模拟的配置与执行流程。核心工作流涉及编写结构化输入脚本,以定义模拟的全局参数、系统构型与动力学设置。该脚本按逻辑划分为明确模块:Initialization、System definition、Settings、Monitoring 与 Run。Initialization 模块建立基础属性,例如单位制(如 units lj)、维度(如 dimension 3)、原子样式(如 atom_style atomic)及边界条件(如 boundary p p p 表示全方向周期性边界)。System definition 模块随后创建模拟盒子(如 create_box)并填充原子(如 create_atoms)。Settings 模块指定原子间势函数(如 pair_style lj/cut 4.0),为原子类型分配质量(如 mass),并定义不同原子类型间相互作用的 Lennard-Jones 参数(如 pair_coeff)。Monitoring 模块配置热力学数据输出(如 thermo_style custom)与可视化输出(如 dump image)。Run 模块包含驱动模拟的命令,例如用于能量最小化的 minimize,以及用于分子动力学的 fix nve 或 fix nvt,并通过 run 命令启动模拟步长。 
该模拟框架基于 LAMMPS 分子动力学引擎构建,按从上到下的顺序依次执行输入脚本中的命令。系统能量与受力基于指定的原子间势函数进行计算,该势函数由 pair_style 与 pair_coeff 命令定义。对于 Lennard-Jones 势,两粒子间的能量由 Eij(r)=4ϵij[(rσij)12−(rσij)6] 给出,其中 r 为粒子间距离,ϵij 为相互作用强度,σij 为势能为零时的距离。LAMMPS 支持多种力场,包括 ReaxFF 与 AIREBO 等多体势,可用于模拟化学键的断裂与形成。模拟的统计系综由 fix 命令控制;例如,fix nve 使用 Velocity-Verlet 算法积分牛顿运动方程,生成微正则(NVE)系综,而 fix nvt 采用 Nosé-Hoover 恒温器维持恒定温度,生成正则(NVT)系综。 
为分析模拟结果,LAMMPS 提供了一套工具集。thermo 命令按指定间隔输出热力学属性(如温度、压力、能量),thermo_style 命令则定义该输出的格式。dump 命令将系统状态(包括原子位置、速度及其他数据)写入文件,以供可视化或后处理使用。dump image 命令通过生成指定时间步的系统图像来辅助可视化。针对更高级的分析,LAMMPS 允许使用 compute 命令计算回转半径或配位数等物理量,这些物理量随后可在 thermo_style 或 dump 命令中调用。fix 命令(如用于巨正则蒙特卡洛的 fix gcmc 或用于反应模拟的 fix bond/react)可用于执行特定采样或模拟化学反应。结果常以 YAML 或 CSV 格式导出,以便在 Python 等外部软件中进行进一步分析。 
该框架同样支持高级模拟技术。例如,fix deform 命令可用于向模拟盒子施加恒定应变率,从而研究材料形变行为。fix addforce 命令可向特定原子组施加恒定外力,从而模拟拉伸聚合物等过程。在计算自由能分布时,fix gcmc 命令可用于巨正则蒙特卡洛模拟,而 fix spring 命令可在伞形采样中沿反应坐标对系统施加偏置力。这些偏置模拟的结果随后可通过加权直方图分析(WHAM)等算法进行处理,以重构无偏自由能分布。 
最后,该框架支持调用外部可视化工具。dump custom 命令可用于输出轨迹文件,该文件可导入 VMD 或 OVITO 等软件中查看。rerun 命令允许在不重新运行模拟的情况下分析轨迹,从而计算初始未设置的物理量。write_data 与 write_restart 命令可将系统状态保存至文件,用于从历史节点恢复模拟或在不同模拟间传输数据。read_data 命令将保存的状态重新读取至 LAMMPS,支持模拟的延续或现有系统的修改。 
实验
基于 LAMMPS 的分子动力学模拟评估了不同系统中的原子弛豫、混合动力学、机械形变与热力学一致性。能量最小化与 Langevin 驱动的混合实验验证了无序初始构型会自然演化为更稳定、能量更低的状态,其特征为跨物种配位数的增加。对比传统力场与反应力场的拉伸形变测试表明,该框架能够准确捕捉弹性拉伸、键断裂及塑性失效过程。此外,自由采样与聚合运行结果证实,该计算框架能够可靠复现预设的热力学势,并在链形成过程中维持稳定的反应动力学。
作者执行了能量最小化模拟,将系统势能由正初始值降至负最终值,表明原子向更稳定的构型迁移。模拟过程追踪了势能随时间的变化,显示初始阶段快速下降,随后逐渐趋近于平台期;由于最小化过程中动能始终为零,系统总能量保持恒定。最小化期间势能由正值降至负值。能量下降初期迅速,随后减缓,最终稳定在负值平台。因最小化全程动能为零,总能量维持不变。
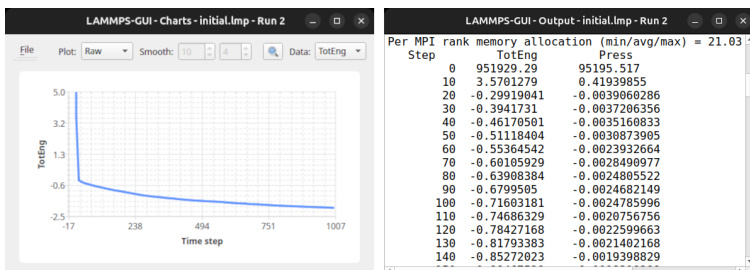
本实验采用能量最小化模拟来评估原子随时间推移的稳定化过程。通过全程追踪势能,结果呈现出初始快速下降随后逐渐趋近于稳定负值平台的过程。这一定性趋势证实,系统在动能为零的前提下成功过渡至低能构型,同时保持总能量恒定。总体而言,该模拟验证了最小化方法在实现结构稳定性方面的有效性。