Command Palette
Search for a command to run...
一键部署 HiDiffusion
摘要
一句话总结
FastRemap 是一款高性能工具,可在不同参考基因组组装版本之间快速重映射基因组测序读数,与成熟的 CrossMap 工具相比,其速度提升最高达 7.82 倍,峰值内存占用降至最低 61.7%。
核心贡献
- FastRemap 被介绍为一种高性能 C++ 实现,旨在对相似参考基因组之间的基因组读数比对进行重映射。
- 该工具修改了核心的 BAM/SAM 重映射逻辑,以解决性能瓶颈,并确保输出结果与标准下游分析流程直接兼容。
- 在人类、秀丽隐杆线虫(C. elegans)和酵母参考基因组上的评估表明,与 CrossMap 相比,其平均速度提升 6.47 倍,平均峰值内存占用仅为 80.7%。
引言
随着基因组数据集的不断扩展以及参考基因组组装版本的频繁更新,研究人员必须高效地将现有的读数比对结果迁移至新的参考基因组。重映射技术利用保守的基因组区域,规避了全量重新比对的计算开销,成为维持高通量基因组分析流程的关键环节。尽管 CrossMap 作为当前标准的重映射工具已被广泛采用,但其仍面临显著的性能瓶颈,且生成的输出格式会破坏与下游变异检测工具及分析软件的兼容性。本文提出 FastRemap,这是一种高度优化的 C++ 实现方案,在加速重映射流程的同时,确保与标准下游工具的即时兼容性。通过对底层算法与数据处理流程的优化,该工具实现了最高 7.82 倍的速度提升,并将峰值内存占用降低至 CrossMap 的约 62%,从而支持更大规模、更快速且更可靠的基因组分析。
方法
作者采用了一种模块化且可扩展的框架,用于将基因组读数从源参考基因组重映射至目标参考基因组。该框架在继承 CrossMap 设计原则的基础上,引入了显著的性能与功能改进。系统的核心在于使用链文件(chain file),该文件定义了源参考基因组与目标参考基因组之间的序列相似区域。此类链文件源自 UCSC 基因组浏览器,且针对每一对参考基因组定制,从而实现精确的坐标转换。该架构设计支持多种数据格式,当前实现主要聚焦于 SAM/BAM 与 BED 文件,未来扩展计划涵盖 VCF、GTF/GFF、BigWig 及 MAF 格式。这种扩展性得益于受 CrossMap 启发的清晰代码库结构。
为实现高效的基因组数据处理,系统依赖 Seqan2 库,该库为序列与比对操作提供了强大的支持。此外,系统集成了 zlib 库以处理压缩文件格式,特别是针对 BAM 输入与输出,从而确保与标准基因组数据处理流程的兼容性。该实现采用 C++ 编写,支持高性能计算,这是其相较于现有工具的关键优势。
如图所示:
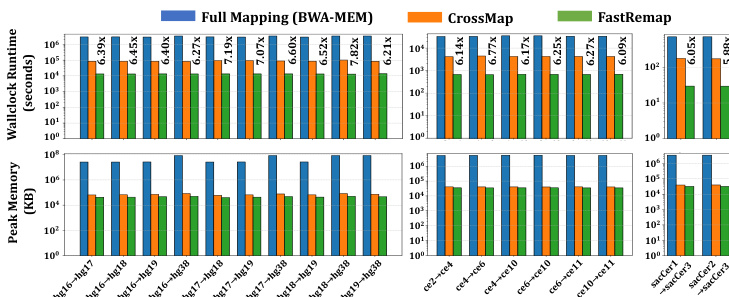
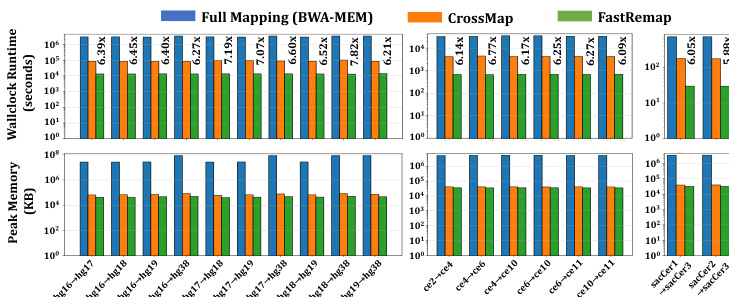 作者从实际运行时间(wall clock runtime)与峰值内存使用量两个维度,评估了 FastRemap 相较于 BWA-MEM 与 CrossMap 的性能。结果表明,FastRemap 相较于 CrossMap 实现了显著的速度提升,尤其在大规模数据集中表现突出,同时保持了较低的内存占用。该性能提升得益于优化的数据结构与高效的链文件遍历算法。
作者从实际运行时间(wall clock runtime)与峰值内存使用量两个维度,评估了 FastRemap 相较于 BWA-MEM 与 CrossMap 的性能。结果表明,FastRemap 相较于 CrossMap 实现了显著的速度提升,尤其在大规模数据集中表现突出,同时保持了较低的内存占用。该性能提升得益于优化的数据结构与高效的链文件遍历算法。
为确保与下游分析流程的兼容性,该系统在原始 CrossMap 实现基础上引入了两项关键改进。首先,FastRemap 在重映射 BAM/SAM 文件时能够正确保留补充比对标志(supplementary alignment flag)。与移除该标志的 CrossMap 不同,FastRemap 保留了准确的比对元数据,使得 Picard 的 MarkDuplicates 等下游工具能够顺利处理输出结果。这确保了重映射后的数据与标准的变异检测及质量控制流程完全兼容。
其次,FastRemap 会生成一个独立的 BED 文件,其中包含未能成功重映射的读数。该输出功能使用户能够快速识别并分析无法比对的读数,而无需将其存储于主 BAM 文件中,从而降低磁盘空间占用并简化后续处理流程。这一设计提升了工具的易用性,并支持大规模测序项目中更高效的数据管理。
实验
该评估在一台高性能服务器上,将 FastRemap 与主流的 CrossMap 工具以及使用 BWA-MEM 进行的全量从头比对(de novo mapping)进行对比,测试范围涵盖多种参考基因组与公开 DNA 测序数据集。通过测量端到端执行时间与峰值内存占用,实验验证了该工具在基因组读数重映射任务中的计算效率与可扩展性。定性结果表明,相较于两种对比方案,FastRemap 提供了显著更快的处理速度与大幅降低的内存需求。最终,该研究确立了 FastRemap 作为现代基因组分析流程中高效且可扩展的解决方案。
作者针对人类、秀丽隐杆线虫与酵母的不同参考基因组配对,将 FastRemap 与 CrossMap 及基于 BWA-MEM 的全量比对进行对比,评估了实际运行时间与峰值内存使用情况。结果显示,FastRemap 的执行速度更快,内存消耗低于 CrossMap 与 BWA-MEM,在速度与内存效率方面均有显著提升。FastRemap 在运行时间与内存使用方面均优于 CrossMap,同时相较于 BWA-MEM 全量比对,显著降低了资源需求。与 CrossMap 相比,FastRemap 实现了更短的实际运行时间与更低的峰值内存占用。相较于 BWA-MEM 全量比对,FastRemap 速度显著更快且内存效率更高。在多种参考基因组配对中,FastRemap 在速度与内存占用方面均优于 CrossMap。
该评估在人类、秀丽隐杆线虫与酵母参考基因组上,将 FastRemap 与 CrossMap 及基于 BWA-MEM 的全量比对进行对比,以评估计算效率。这些实验通过测量执行速度与内存消耗,验证了各工具的实际资源需求。总体而言,研究结果证实 FastRemap 能够持续提供优越的运行性能与更低的内存开销,使其成为跨基因组映射任务中更为高效的解决方案。