Command Palette
Search for a command to run...
VASP在GPU上的应用:用于计算元素硼稳定性的精确交换
VASP在GPU上的应用:用于计算元素硼稳定性的精确交换
M. Hutchinson M. Widom
VASP 结合 phonopy 计算硅的声子谱
摘要
通用图形处理单元(GPU)为某些高度可并行的计算任务提供了极高的处理速度,例如矩阵运算和傅里叶变换,而这些任务是第一性原理电子结构计算的核心。引入精确交换会使密度泛函理论的计算成本增加几个数量级,从而促使人们使用GPU。将广泛使用的电子密度泛函代码VASP移植到GPU上运行,使得精确交换的计算性能相比传统CPU提升了5-20倍。我们分析了性能瓶颈,并讨论了将从GPU中受益的问题类别。作为该实现能力的一个示例,我们利用精确交换计算了α和β菱形硼结构的晶格稳定性。我们的结果证实了在低温下β菱形结构因对称性破缺的部分占据而具有能量优势,但未能确定α相对于β的稳定性。
一句话总结
将VASP密度泛函理论代码移植到GPU架构上,可使精确交换计算的性能较CPU实现提升5至20倍。这一结论通过元素硼在低温下的晶格稳定性评估得到验证,该评估证实了β-菱形相中存在破坏对称性的部分占据现象,但尚未明确其相对于α结构的稳定性。
核心贡献
- 本文提出了一种基于GPU加速的VASP电子结构代码移植版本,能够高效计算精确交换项,而该项传统上会使密度泛函理论的计算成本增加数个数量级。该实现利用了矩阵运算和傅里叶变换的高并行性,实现了较传统CPU执行5至20倍的性能提升。
- 分析了精确交换例程中的计算瓶颈,以识别最受益于GPU加速的特定电子结构问题类别。此项评估为扩展并行化量子化学工作流的优化策略奠定了基础。
- 该加速框架使用HSE06杂化泛函计算了α型和β型菱形硼多晶型的相对晶格稳定性。基准测试结果证实,在低温下能量上更倾向于β型菱形结构的破坏对称性的部分占据。
引言
第一性原理量子力学模拟对于预测材料性能至关重要,但随着体系规模的扩大,会面临严重的计算瓶颈。确定硼等复杂元素稳定晶体结构需要极高精度的电子结构方法,而标准密度泛函理论近似无法解析竞争多晶型之间微小的能量差异。包含精确电子交换的更高精度杂化泛函在传统处理器上计算成本过高,历史上仅能应用于较小体系。本文作者利用GPU加速技术移植VASP代码库,专门针对大规模并行性优化了精确交换计算。该方法较CPU实现了高达二十倍的性能提升,同时支持进行精确的能量比较,从而解决了关于硼结构稳定性的长期争议。
数据集
- 数据集组成与来源: 作者利用VASP软件包通过第一性原理密度泛函理论(DFT)计算,汇编了包含四种晶体硼相的基准测试集。
- 子集详情: 该集合包含一个12原子的α-菱形结构(hR12)、一个通过加倍晶格轴以匹配β参数构建的96原子α超胞(hR12x8)、一个105原子的理想β-菱形结构(hR105),以及一个针对GGA总能量优化的107原子对称性破缺β变体(aP107)。
- 数据使用与处理: 作者利用这些结构对传统DFT与混合HF-DFT方法之间的计算效率和能量收敛性进行基准测试。为高效完成基准测试,优先采用2x2x2 k点网格,同时保留3x3x3网格用于高精度收敛测试,以解析meV/atom级别的能量差异。
- 网格与配置: 根据结构对称性和晶格缩放调整Monkhorst-Pack k点网格,实空间FFT网格随晶格尺寸成比例缩放。所有计算均采用投影缀加平面波(PAW)势、PBE交换关联泛函、固定的319 eV平面波截断能以及PREC=Normal设置,以确保电荷密度表示的准确性并避免边界包裹误差。
方法
作者利用基于GPU加速的VASP代码实现来进行电子结构模拟的精确交换计算,重点提升涉及元素硼等高要求任务的性能。该框架旨在将GPU加速集成到现有VASP架构中,同时保持其模块化和基于抽象的结构。实现过程涉及在五个关键节点拦截主执行流:初始化和清理例程、用于大规模变换的FFT3D例程,以及FOCK_ACC和FOCK_FORCE例程。这些拦截点允许将计算密集型操作选择性卸载至GPU,而较小或计算量较低的任务则保留在CPU上。
精确交换计算的核心计算任务涉及一个四层嵌套循环结构:外层两个循环遍历k点索引,内层两个循环遍历能带索引,计算对Fock交换能的贡献。作者通过将内层循环(特别是与能带相关的操作)卸载至GPU来优化此过程。该策略将矩阵-向量运算转换为效率更高的矩阵-矩阵运算,从而实现更高的并行度并更好地利用GPU资源。GPU上的内存占用通过NSIM参数进行动态控制,该参数管理并发处理的能带数量。为最小化主机与设备间的数据传输开销,循环内的非密集型操作也在GPU上执行。
该实现分别依赖cuFFT和cuBLAS库来执行高性能FFT和BLAS运算。此外,开发了20个自定义CUDA内核以复现CPU功能,确保与VASP现有计算逻辑兼容。通过CUDA流实现异步执行,支持较小内核的并发运行并提升性能,尤其适用于较小的输入结构。该框架同时支持全双精度和混合精度算术,两种设置间的数值差异微乎其微(在千分之一百分比以内),证明了其高度的准确性。

实验
本研究通过在多种硼晶体结构上对双GPU工作站与多核CPU超级计算机配置进行基准测试,评估了GPU加速精确交换密度泛函理论的计算性能与物理准确性。性能实验验证了GPU加速能有效规避CPU通信瓶颈,其运行时间与大规模集群运行相当,使以往难以实现的杂化泛函计算在桌面硬件上成为可能。同时,结构能量学分析表明,精确交换方法在特定β多晶型上更倾向于α-硼,且不同泛函下的能量收敛模式表明,仍需更高级的理论方法才能最终确定硼的稳定低温相。总体而言,这些结果确立了基于GPU的加速作为一种可扩展且具成本效益的策略,可将精确交换模拟的应用扩展至更大的材料体系和动态过程。
作者分析了硼结构GPU加速计算的性能,比较了不同配置和平台下的运行时组件。结果显示,GPU实现在特定计算例程中显著提速,其性能扩展效果优于基于CPU的系统,尤其在通信开销有限的较小结构中。与仅使用CPU的实现相比,GPU加速在关键计算例程中提供了巨大的速度提升。由于GPU系统通信开销降低,性能增益在较小结构中最为显著。GPU系统达到了与多核CPU配置相当的性能,使桌面硬件能够进行高级计算。
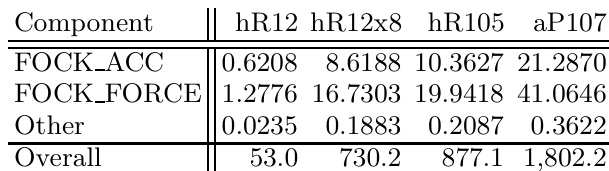
作者比较了使用VASP结合杂化泛函计算硼结构能量时GPU与CPU系统的性能。结果表明,GPU实现显著优于单核CPU,使原本需要大型超级计算资源的计算得以在桌面硬件上完成。GPU的性能优势在较大结构中尤为明显,此时GPU系统的求解时间与多核CPU配置相当或更优。GPU加速使桌面硬件能够进行原本需要超级计算资源的精确交换计算。对于大型结构,GPU系统达到了与超级计算机上众多CPU核心相当的性能。GPU的性能优势随结构尺寸增加而提升,使其在更大体系中更为有效。
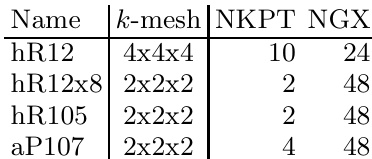
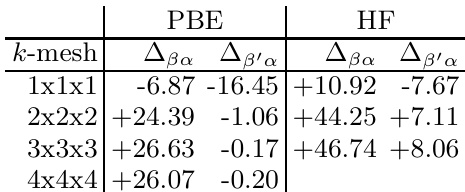
作者展示了使用不同密度泛函计算的结构能量对比,表明PBE与HF方法之间存在显著差异。结果表明,HF方法对相同结构给出更高的能量值,说明交换关联泛函对预测硼结构稳定性具有显著影响。在所有测试构型中,HF方法预测的结构能量均高于PBE方法。PBE与HF之间的能量差异在某些k网格下更为明显,表明对计算参数具有敏感性。该对比突显了泛函选择在确定硼结构相对稳定性方面的重要性。
作者比较了不同密度泛函下的结构能量,显示β结构的能量高于α结构,且hR141结构的优化变体降低了每个原子的能量。结果表明能量向局域密度近似值收敛,说明需要更高级的理论来解析硼结构的稳定性。在所有泛函下,β结构的能量均高于α结构。hR141结构的优化变体每个原子的能量低于理想β结构。能量值向局域密度近似收敛,而非广义梯度近似。
作者比较了CPU与GPU实现在计算硼结构能量时的性能,重点关注运行时间和计算效率。结果表明,GPU实现显著优于单核CPU,使以前在标准硬件上难以研究的复杂结构得以探索。GPU在关键计算例程中实现大幅提速,使高级计算可在桌面系统上运行。GPU实现在单核CPU基础上实现显著加速,使以往难以实现的计算得以在桌面硬件上进行。GPU性能显著快于纯CPU系统,计算效率最高提升一个数量级。GPU的高性能使得研究原本需要超级计算资源的复杂结构成为可能。
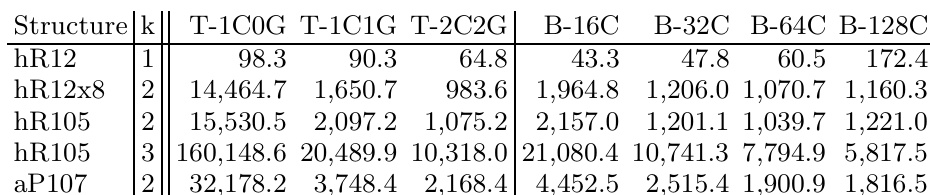
本研究评估了硼结构的GPU加速密度泛函理论计算,比较了跨平台的硬件性能,并验证了交换关联泛函对预测稳定性的影响。基准测试表明,GPU实现在单核CPU基础上提供了显著的计算加速,使高级杂化泛函计算在桌面系统上成为可能,且性能优势在较大结构中呈现良好的扩展性。泛函对比显示,杂化方法一致给出比梯度近似更高的结构能量,且结果对计算参数高度敏感。此外,针对特定硼多晶型的跨泛函分析表明,β相不如α相稳定,而优化结构向局域密度近似极限收敛,这凸显了需要先进理论方法来最终确定硼相稳定性的必要性。