Command Palette
Search for a command to run...
基于 220 种海洋细菌,科学家用基因组尺度模型重构异养微生物分类体系,挖出 8 类代谢菌群

森林被誉为地球之肺,海洋则是地球之心。广袤大洋中数以万计的微生物组成复杂群落,依靠各自独特的代谢分工调控有机质转化,驱动碳固定与碳释放过程,深刻左右全球碳循环、气候变化与海洋生物多样性。其中,海洋异养微生物犹如海洋生态系统中的「净化单元」,承担着有机质降解的核心功能,从而维系全球物质循环与生态平衡。
长久以来,海洋异养微生物被经典理论划分为富营养型(copiotrophic)与寡营养型(oligotrophic)两大类,前者在高有机质环境旺盛生长,后者在资源贫乏环境低速存活。这套沿用多年的传统「二分法」虽在一定程度上助力了生物地球化学的研究,但也存在显著短板:生长快慢无法等同于底物利用偏好与代谢生态位,如同不能仅凭饭量划分人类饮食习惯一般,「爱吃什么」才是决定有机质分解速率、调控碳循环走向的关键。
针对于此,南加利福尼亚大学牵头的团队依托全球海洋微生物数据库(Ocean Microbial Database,OMD),借助基因组尺度代谢模型 Genome-scale metabolic models(GEMs)解析海量海洋细菌基因组,通过定量微生物对 11 类有机底物的利用敏感性,最终打破传统「二分法」框架,划分出 8 类差异化代谢菌群:1 类速生富营养型菌群、 3 类底物专一的慢生寡营养型菌群、 4 类底物特化的中间生长型菌群。
相关成果以「Defining metabolic niches for marine microbial heterotrophs」为题,发表于 Science Advances 。
研究亮点:
* 突破经典「二分法」框架,以实际代谢策略、底物偏好锚定微生物专属代谢生态位
* 依托 8 类功能菌群,系统揭示海洋异养微生物的生长规律、资源竞争模式与全球地理分布,解析了微生物驱动海洋碳循环的内在机制
* 补齐海洋异养微生物参与全球碳循环的研究短板,为生物地球化学模型提供精细化改良思路与参数方案

论文地址:
https://www.science.org/doi/10.1126/sciadv.adz0537
数据集:涵盖 220 个不同类目的海洋细菌
本研究依托于一项大型海洋微生物基因组数据,数据集取自托管于 microbiomics.io 平台的 OMD 数据库。该数据库收录约 35,000 个微生物基因组,包含宏基因组组装基因组、单细胞扩增基因组与人工分离培养菌株基因组。
本次研究中仅纳入完整度 > 80% 、污染率 < 5% 的细菌基因组。两组数值分别通过 CheckM 和 Anvi’o 两款软件的评分均值核算得出。随后,研究人员使用 OMD 数据库附带的元数据,采用 dRep 软件以 95% 平均核苷酸一致性(Average Nucleotide Identity,ANI)为阈值对基因组去冗余。
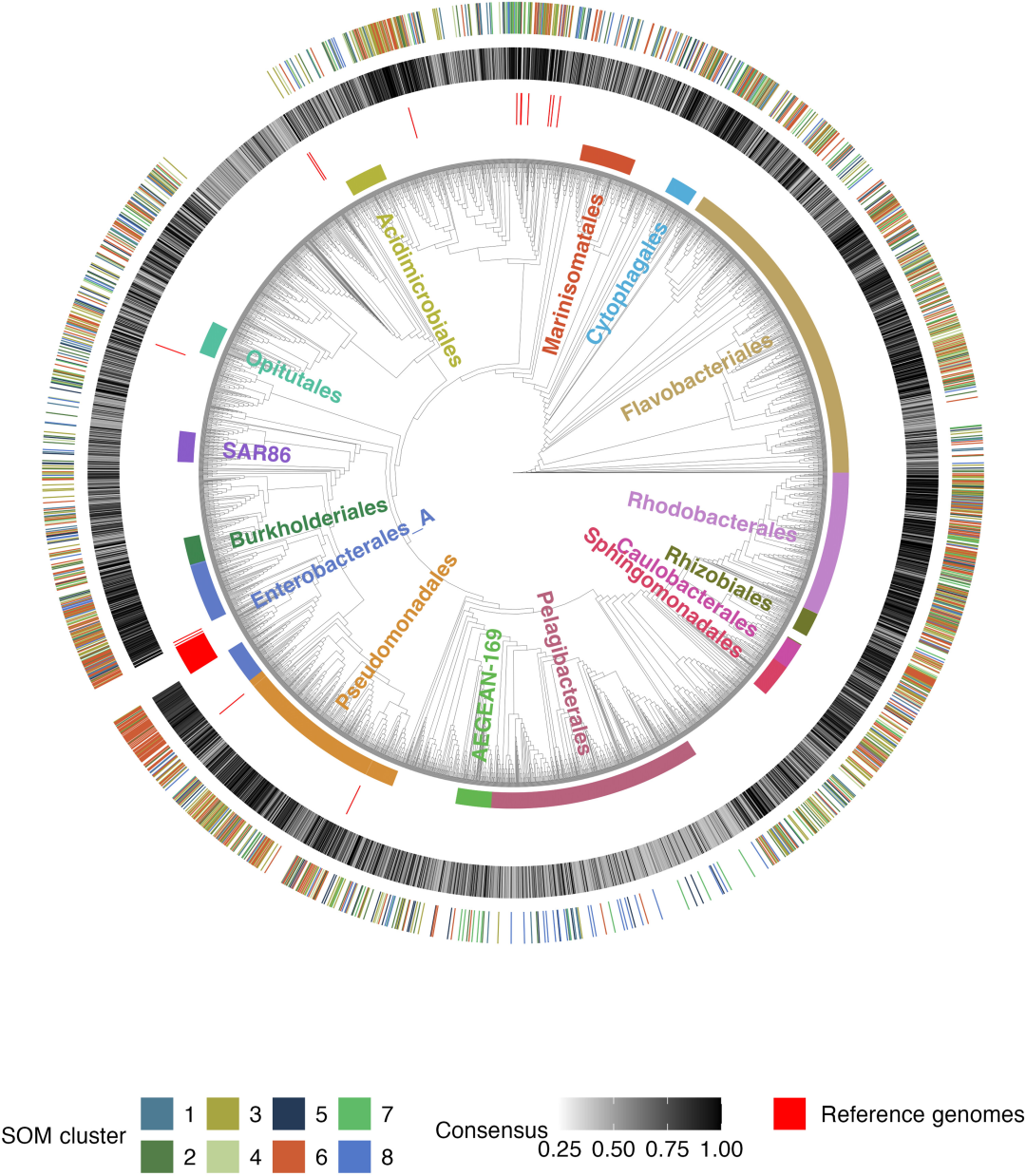
在剔除 180 株光合自养型蓝藻菌(外类群)后,最终筛选得到 3,738 个经过去冗余处理的高质量异养细菌基因组,构成了本次研究的基础分析数据集。数据集涵盖 220 个不同类目的海洋细菌,其中 14 个类群所含基因组数量 ≥ 50 。
另外在系统发育树(Phylogenetic tree)的构建中,研究除保留 180 株蓝藻菌基因组作为外类群外,额外加入 BiGG 数据库 66 株细菌参考基因组,基因组总数合计达到 3,984 株。其中,8 个基因组因目标单拷贝基因匹配不足剔除出发育树,最终 3,976 株用于构建发育树,并依托 GTDB-Tk v2.1.0 和 GTDB r214 数据库在树上标注全部基因组分类信息。
自组织映射神经网络划分代谢生态位
为了突破传统「二分法」框架的局限,本研究整合了基因组学、约束性代谢模拟与无监督机器学习技术,搭建了一套从基因信息到菌群生态分型的完整分析框架,通过多类实测与全球环境数据集,分层完成代谢建模、底物敏感性量化与菌群聚类划分。
建模与质量把控
在模型构建环节,研究人员以集成建模策略,采用 CarveMe v1.5.1 软件为 3738 株海洋异养细菌各自构建 60 组独立代谢模型(模型集合)。
具体来说,CarveMe 软件的建模原理是依托通用代谢模型架构,根据输入基因组注释信息中各生化反应的存在与否,为每步反应赋予权重,以此完成通用模型初始化,进而预测对应基因组的代谢模型。本研究全面探索了覆盖单个基因组反应谱所需的模型重复数,结果显示当集合内模型数量达到 60 左右时,新增反应的总量区域平稳,说明 60 个模型组成的集合即可覆盖单个基因组绝大多数可行代谢模型组合。
为量化 CarveMe 输出代谢模型的优劣,研究还首创了一致性得分指标 C 作为评估,公式如下:
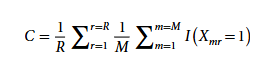
公式中,Xmr 代表集成模型反应在 M 个独立构建模型中的「存在-缺失矩阵」;r 指代单个生化反应;R 为整套模型集合中全部反应的总数;I 为指示函数,用于判定反应 r 是否存在于第 m 个集成子模型中。后续分析仅保留一致性得分 ≥ 0.8 的基因组样本,共计 1,578 组基因组。
代谢策略评估
研究人员将代谢策略定义为生物体生长时首选的一组底物,研究方法通过一系列敏感性分析来解释。
具体来说,研究人员借助 COBRApy v0.25.0 软件包中的通量平衡分析(FBA)工具包,在「底物充足」和「底物受限」两种底物供给条件下完成 CarveMe 模型的生长敏感性测试。「 底物受限」条件的设置方式为:将某一类化合物的可用通量缩减至该生物在「底物充足」条件下摄取量的 50% 。
为量化不同模型间底物需求的差异性,研究提出一个敏感性系数指标 S,如下所示:
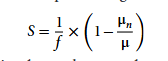
公式中,μn 代表第 n 类底物受限条件下的预测生长速率,μ 为底物充足条件下的预测生长速率,f 为底物限制系数(研究取 0.5)。敏感性系数 S 取值范围为 [0,1],表示若底物供给量下调 50% 后,模型测算的生长速率同步下降 50%,则判定该类底物是生物生长的完全限制因子(生长敏感度为 1);若模型生长速率未出现任何变化,则说明该生物的生长不受此类底物供给量影响(生长敏感度为 0)。
另外,当底物受限幅度与生长速率下降幅度比值 ≥ 0.8(S ≥ 0.8)时,则判定该模型对此类底物具备显著生长敏感性。
无监督机器学习的聚类分析
研究的机器学习部分采用了自组织映射神经网络(Self-Organizing Maps ,SOMs)划分代谢生态位。 SOMs 是一种无监督机器学习算法, 可将海量高维数据集降维至具备拓扑结构的二维网格空间。
聚类前,研究人员对前述得到的 1,578 个基因组增设了一轮数据筛选,统计全部 60 个子模型中各类代谢物对应的生长敏感方差,剔除了 100 组底物敏感度总方差 > 0.1 的基因组,最终剩余 1,478 个基因组,共计 88,680 组(1,478 个基因组 x 60 个集成模型)有效数据用于 SOMs 聚类分析,每条数据包含 11 项代谢敏感度特征指标。
具体设置上,本研究采用 kohonen v3.0.12 软件对标准化后的化合物通量预测数据进行处理,在 20 x 20 尺寸的环面六边形网格(采用标准欧氏距离表征空间距离)上完成 1,500 次迭代运算,学习率参数设为(0.025,0.01),邻域半径选用软件默认值。
经过充分训练后,基于生长化合物敏感性预测结果的一致性,采用 K 均值聚类(K-means clustering)算法,最终将 SOMs 图谱划分为 8 个差异化聚类。
聚类完成后,为评估最大生长速率的差异,研究通过 gRodon 计算了所有 1478 组基因组的 dCUB,从而划分出快慢生长类型,并依托 Tara Oceans 、 BioGeoTraces 和 Malaspina 数个数据集的 1,209 个宏基因组,以及 McNichol 等人的 一项全球扩增子序列变体数据集(Global ASV Dataset)完成了 8 类菌群的全球地理分布验证。
以底物偏好+生长速率划分 8 类代谢集群
研究针对性展示了多样化的实验结果,不仅验证了模型性能,更重要的是突破了传统「二分法」框架,提出一套全新的分类逻辑,构建起底物偏好与代谢生态位的内在关联。
模型验证结果
研究人员通过对 186 种海洋微生物的碳源偏好进行大规模培养实验分析,验证了 CarveMe 模型捕捉实验观测到底物偏好能力的准确性。具体来看,研究人员为 Gralka 等人研究中的基因组构建了 CarveMe 模型,并利用 FBA 进行了对应的计算机模拟实验(in silico experiment),测试这些微生物在相同碳源条件下的生长情况。
结果显示,模型预测结果与文献中的实验数据相比,达到了 75.5% 的准确率(accuracy)和估计为 87.4% 的精确率(precision)。为了评估这一结果是否显著优于随机预测,研究人员通过对随机预测进行自助法(bootstrap)分析进行了检验,结果表明模型的准确率显著高于随机水平。
8 类菌群分型结果
依托 1,478 组基因组、 11 项敏感指标,研究通过 SOMs 聚类得到 8 类差异化代谢菌群,并按照生长速率的不同将其划分为快、中、慢三大类,具体来看(如下图所示):
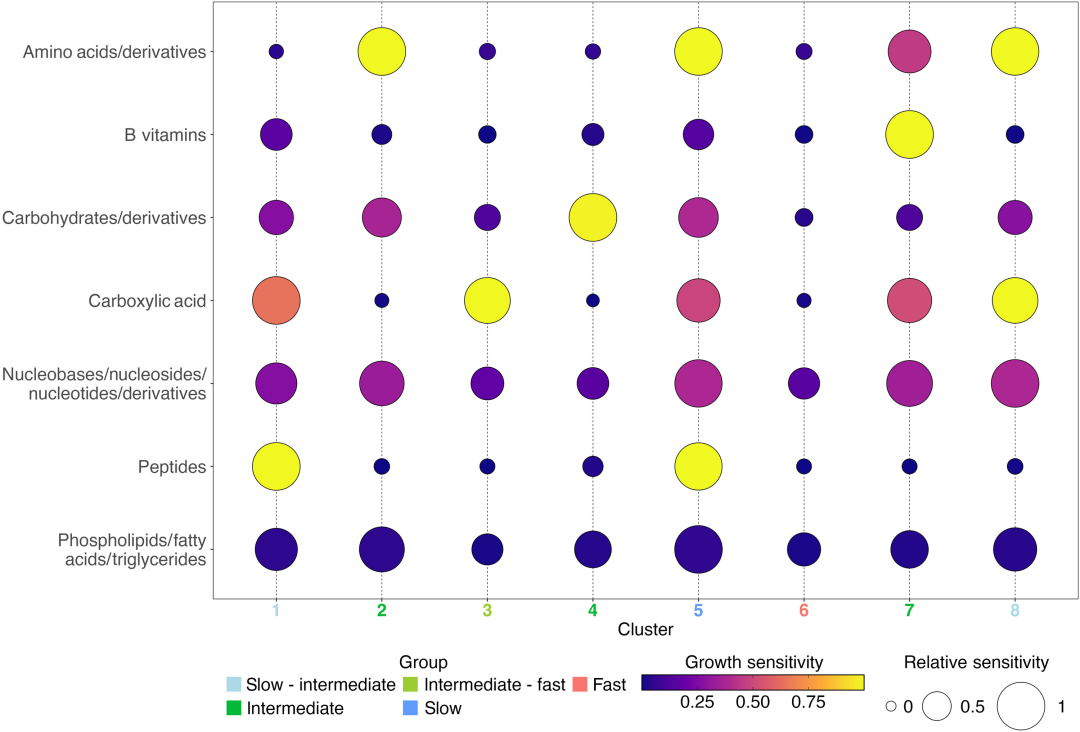
8 个 SOM 聚类的平均生长敏感性对比
1 类速生富营养型菌群(集群 6):这是典型的富营养型菌群,其中 79.5% 的基因组预测最大基因组生长速率高于慢速生长临界预测值(dCUB <-0.08 为快速生长临界值,dCUB 数值越小代表生长速度越快)。从分类学角度来看,集群 6 的典型代表有 Enterobacterales 、 Flavobacteriales 、 Rhodobacterales 和 Pseudomonadale,此类菌群受到底物影响程度最低,在受试的 11 类化合物中,缺失任意一类都不会抑制其生长。
3 类底物专一的慢生寡营养型菌群(集群 1 、集群 5 和集群 8):此类菌群 dCUB = -0.111 。其中,集群 5(占 61.8%)最大生长速率最低,典型代表有 Opitutales (Verrucomicrobiota 门) 和 Pelagibacterales,两类菌群在本群中的富集度分别达到 435% 和 362% 。
4 类底物特化的中间生长型菌群(集群 2 、集群 3 、集群 4 和集群 7):这类菌群预测生长速率显著低于集群 6,但明显高于集群 5 。其中集群 3 的生长速率还明显优于集群 1 、 8 。这四类中间生长型菌群生长速率类群各自仅对一类化合物表现出生长响应敏感性,分别是氨基酸、羧酸、碳水化合物与 B 族维生素。
另外,中间生长型菌群的特征也验证了近期一项研究,海洋次表层环境中的优势异养微生物类群或为慢速生长的富营养菌。该结果可为这类微生物划分代谢功能类群提供依据,比如集群 4 偏好糖类。
写在最后
总的来说,本次研究跳出了沿用数十年的富营养型/寡营养型「二分法」框架,从基因与底物利用本质出发,建立了 8 类代谢生态位分类体系,打破了五种分类与生理功能的固有绑定认知。
此外,这套全新分类框架简化了海洋海量异养微生物的复杂结构,未来可以被嵌入全球生物地球化学模型,不再需要逐一录入上万种海洋细菌,仅依靠 8 类功能参数即可推演海洋有机质降解、碳收支变化,为研判全球变暖背景下大洋碳循环演变提供了全新的理论工具。








