Command Palette
Search for a command to run...
OpenTME: مجموعة بيانات مفتوحة تحتوي على ملفات تعريف للميكروبيوم الورمي المُستخلصة من تقنية H&E باستخدام الذكاء الاصطناعي، ومستمدة من مشروع TCGA.
OpenTME: مجموعة بيانات مفتوحة تحتوي على ملفات تعريف للميكروبيوم الورمي المُستخلصة من تقنية H&E باستخدام الذكاء الاصطناعي، ومستمدة من مشروع TCGA.
الملخص
يلعب البيئة الدقيقة للورم (TME) دوراً محورياً في تطور السرطان، والاستجابة للعلاج، والنتائج الصحية للمرضى؛ ومع ذلك، لا تزال هناك ندرة في وجود بيانات واسعة النطاق ومتسقة وكمية لوصف البيئة الدقيقة للورم مشتقة من الصور النسيجية التقليدية الملونة بالهيماتوكسيلين واليوزين (H&E). نقدم في هذا العمل "OpenTME"، وهو مجموعة بيانات مفتوحة الوصول تحتوي على ملفات وصفية مسبقة الحساب للبيئة الدقيقة للورم، مستمدة من 3,634 صورة شريحة كاملة (Whole-Slide Images) ملونة بـ H&E، تغطي خمسة أنواع من السرطان (سرطان المثانة، والثدي، والقولون والمستقيم، والكبد، والرئة)، ومستمدة من مشروع خريطة الجينوم السرطاني (The Cancer Genome Atlas - TCGA).تم إنشاء جميع المخرجات باستخدام "Atlas H&E-TME"، وهو تطبيق يعمل بالذكاء الاصطناعي ومبني على عائلة نماذج الأساس في علم الأمراض (Pathology Foundation Models) الخاصة بـ Atlas. يقوم التطبيق بتنفيذ مراقبة جودة النسيج، وتجزئة النسيج، واكتشاف الخلايا وتصنيفها، وتحليل الجوار المكاني، مما ينتج عنه أكثر من 4,500 مقياس كمي لكل شريحة بدقة على مستوى الخلية. يمكن الوصول إلى "OpenTME" لأغراض البحث الأكاديمي غير التجاري عبر منصة Hugging Face.نحن ملتزمون بتوسيع نطاق "OpenTME" على مر الزمن، ونتوقع أن يصبح موردًا قيمًا لاكتشاف المؤشرات الحيوية، وأبحاث البيولوجيا المكانية، وتطوير الأساليب الحسابية لتحليل البيئة الدقيقة للورم.
One-sentence Summary
To address the scarcity of large-scale, consistent, and quantitative tumor microenvironment characterization from routine H&E-stained histopathology, the paper introduce OpenTME, an open-access dataset of pre-computed TME profiles from 3,634 H&E-stained whole-slide images across five cancer types (bladder, breast, colorectal, liver, and lung) from The Cancer Genome Atlas (TCGA), generated using Atlas H&E-TME, an AI-powered application built on the Atlas family of pathology foundation models that performs tissue quality control, tissue segmentation, cell detection and classification, and spatial neighborhood analysis, yielding over 4,500 quantitative readouts per slide at cell-level resolution, and available on Hugging Face for non-commercial academic research to support biomarker discovery, spatial biology research, and the development of computational methods for TME analysis.
Key Contributions
- OpenTME is an open-access dataset of pre-computed tumor microenvironment profiles derived from 3,634 H&E-stained whole-slide images across five cancer types (bladder, breast, colorectal, liver, and lung cancer) from The Cancer Genome Atlas (TCGA), providing over 4,500 quantitative readouts per slide at cell-level resolution.
- The profiles were generated using Atlas H&E-TME, an AI-powered application built on the Atlas family of pathology foundation models that performs tissue quality control, tissue segmentation, cell detection and classification, and spatial neighborhood analysis.
- The dataset is available on Hugging Face for non-commercial academic research and is anticipated to serve as a resource for biomarker discovery, spatial biology research, and the development of computational methods for TME analysis.
Introduction
The tumor microenvironment (TME) captured in routine H&E-stained histology slides holds critical prognostic and predictive information, yet systematic extraction of TME features from these images has been hindered by manual annotation bottlenecks and limited reproducibility. Although The Cancer Genome Atlas (TCGA) offers a wealth of digitized H&E slides, publicly available, AI-derived TME profiles at scale have been missing, forcing researchers to rerun costly inference pipelines or rely on small-scale annotations. The authors address this gap with OpenTME, an open dataset of AI-powered TME profiles computed from TCGA H&E whole-slide images, giving the community ready-to-use, standardized features that accelerate computational pathology research and enable robust benchmarking.
Dataset
The authors introduce OpenTME, a ready-to-use dataset of quantitative tumor microenvironment profiles derived from routine H&E-stained whole-slide images. It compiles pre-computed outputs from the AI-powered Atlas H&E-TME pipeline, which applies tissue quality control, tissue segmentation, cell detection and classification, and spatial neighborhood analysis to each slide.
- Data source: Diagnostic formalin-fixed paraffin-embedded (FFPE) slides from The Cancer Genome Atlas (TCGA), accessed via the NCI Genomic Data Commons.
- Cancer types and projects: Five indications across eight TCGA projects: bladder, breast, colorectal, liver, and lung cancer.
- Final dataset size: 3,634 slides, after excluding 52 slides from an initial set of 3,686. Exclusions: 49 due to missing resolution metadata or file corruption, 2 non‑H&E stains (1 IHC, 1 Masson’s trichrome), and 1 slide that failed quality control because of fully out‑of‑focus tissue.
- What each slide includes:
- Over 4,500 quantitative readouts in CSV format, grouped into slide‑level tables per cancer type. The features span:
- Tissue QC metrics (area and relative coverage per QC region)
- Tissue segmentation metrics (area, count, roundness, eccentricity, etc., for seven tissue types)
- Cell metrics (count, percentage, density, nuclear morphology per nine cell types, both slide‑level and stratified by tissue compartment)
- Neighborhood metrics (spatial co‑occurrence statistics, ratios, densities within 20 µm and 40 µm radii)
- Thumbnail images with overlay visualizations of tissue QC, tissue segmentation, and cell classification predictions.
- Over 4,500 quantitative readouts in CSV format, grouped into slide‑level tables per cancer type. The features span:
- How the dataset is used: The paper provides OpenTME as a resource for downstream research without requiring users to run AI inference. TME Studio, a collection of interactive marimo notebooks, accompanies the dataset with tutorials, immune infiltrate classification examples, Kaplan–Meier survival analysis, and visualizations. The authors intend it for biomarker discovery, spatial biology studies, and development of new computational pathology methods. No training split or mixture ratios apply, as the dataset contains aggregated slide‑level features, not raw images.
- Additional processing notes: All features are generated by the Atlas H&E-TME application, which runs tissue quality control, segmentation into seven tissue types, cell detection and classification into nine cell types, and spatial neighborhood analysis. No patch‑level cropping is used; features are aggregated at the slide level. Researchers who need spatially resolved outputs (cell coordinates, polygon geometries) can apply through the Atlas H&E-TME Research Access Program.
- Access and restrictions: The dataset is available on Hugging Face under a gated access model for non‑commercial academic research. Training models to replicate Atlas H&E-TME capabilities is prohibited, and users must comply with TCGA data use policies.
Method
The authors leverage a multi-stagecomputational pipeline within the Atlas H&E-TME application to process Whole Slide Images (WSIs) and extract detailed tumor microenvironment features. This pipeline consists of three sequential deep learning models: Tissue Quality Control, Tissue Segmentation, and Cell Classification.
As shown in the figure below:
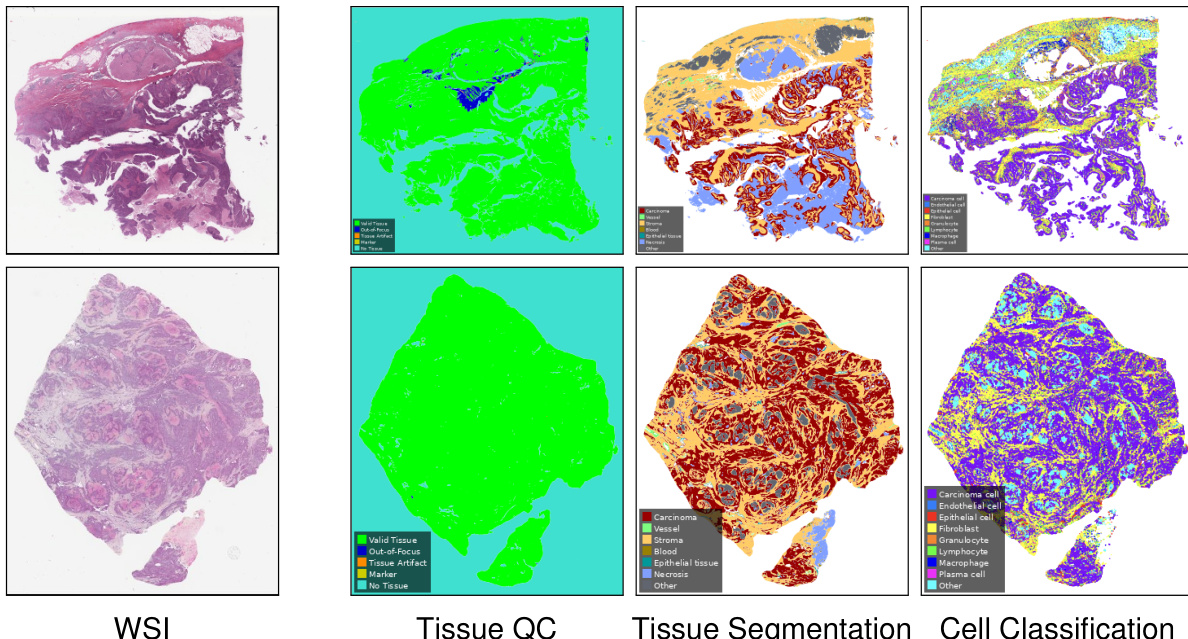
The process begins with the Tissue QC model, which evaluates the input WSI to identify valid tissue regions, filter out artifacts, and exclude out-of-focus areas or markers. This step ensures that downstream analyses are performed only on high-quality tissue data. Following the quality control step, the Tissue Segmentation model partitions the valid tissue into distinct histological compartments. This model classifies regions into categories such as carcinoma, stroma, blood, epithelial tissue, and necrosis.
Finally, the Cell Classification model operates on the segmented tissue to identify and categorize individual cells within the tumor microenvironment. This model distinguishes between various cell types, including carcinoma cells, endothelial cells, epithelial cells, fibroblasts, granulocytes, lymphocytes, macrophages, and plasma cells. By chaining these three models, the application generates comprehensive slide-level tissue and cell readouts, as well as neighborhood readouts, enabling a granular analysis of the tissue architecture.
Experiment
The evaluation setup involved validating the Atlas H&E-TME application—a four-stage AI pipeline for single-cell tumor microenvironment profiling in H&E slides—against annotations from board-certified pathologists using diverse multi-source datasets and scanner types across five cancer indications. The validation confirmed that the system reliably performs tissue quality control, segments seven tissue classes, detects and classifies cells into nine types, and derives spatial readouts such as cell densities and neighborhood co-occurrence statistics. The overall findings demonstrate robust coverage of at least 90% of invasive morphological subtypes per supported cancer, underscoring the model's generalizability and practical utility for comprehensive spatial profiling in clinical research.