Command Palette
Search for a command to run...
مجموعة من الدروس التعليمية لحزمة محاكاة LAMMPS
مجموعة من الدروس التعليمية لحزمة محاكاة LAMMPS
Simon Gravelle Cecilia M. S. Alvaras Jacob R. Gissinger Axel Kohlmeyer
نشر لمرة واحدة لدليل LAMMPS للمبتدئين: تقدير نقطة انصهار FCC Cu في نظام NPT
الملخص
يتيح توفر حزم برمجيات المحاكاة الجزيئية مفتوحة المصدر للعلماء والمهندسين التركيز على تشغيل المحاكاة وتحليلها دون الحاجة إلى كتابة برمجيات المحاكاة الخاصة بهم، أو موازنتها، أو التحقق من صحتها. ومع ذلك، فإن الطبيعة "الصندوق الأسود" لهذه الحزم البرمجية، والتنوع الواسع للخيارات والميزات المتاحة، قد يجعل استخدامها بشكل صحيح أمراً صعباً، لا سيما للمبتدئين في مجال المحاكاة. يُعد LAMMPS أحد أكواد المحاكاة الجزيئية متعددة الاستخدامات، والمصمم لنمذجة الأنظمة القائمة على الجسيمات عبر طيف واسع من تطبيقات علوم المواد والكيمياء الحاسوبية، بما في ذلك النماذج الذرية، والمبسطة (coarse-grained)، والمتوسطة المقياس (mesoscale)، والمستمر الخالي من الشبكات (grid-free continuum)، والعناصر المنفصلة. ويتميز LAMMPS بقدرته على تشغيل المحاكاة بكفاءة بأحجام متفاوتة، بدءاً من أجهزة الحاسوب المكتبية الصغيرة وصولاً إلى بيئات الحوسبة الفائقة واسعة النطاق. وتُعد مرونته وقابليته للتوسعة مثالية لإجراء محاكيات معقدة وموسعة للأنظمة الذرية والجزيئية وما وراءها. تقدم هذه المقالة مجموعة من الدروس التعليمية المصممة لتسهيل تعلم LAMMPS للمستخدمين الجدد. تغطي الدروس الأربعة الأولى أساسيات تشغيل المحاكاة الجزيئية في LAMMPS باستخدام أنظمة ذات تعقيدات متفاوتة. بينما تتناول الدروس الأربعة التالية تقنيات أكثر تقدماً في المحاكاة الجزيئية، وتحديداً استخدام مجال القوة التفاعلي (reactive force field)، ومونت كارلو الكنسي الكبير (grand canonical Monte Carlo)، وأخذ العينات المحسّن (enhanced sampling)، وبروتوكول REACTER. بالإضافة إلى ذلك، نقدم LAMMPS-GUI، وهو محرر نصوص رسامي محسّن متعدد المنصات، مصمم خصيصاً للاستخدام مع LAMMPS وقادر على تشغيل LAMMPS مباشرةً على الإدخال المُحرَّر. يُستخدم LAMMPS-GUI كأداة رئيسية في الدروس التعليمية لتحرير المدخلات، وتشغيل LAMMPS، واستخراج البيانات، وتصوير الأنظمة المُحاكاة.
One-sentence Summary
This article introduces a structured tutorial suite and LAMMPS-GUI, a cross-platform graphical text editor that consolidates input editing, simulation execution, data extraction, and visualization, guiding users from foundational setups to advanced techniques such as reactive force fields, grand canonical Monte Carlo, enhanced sampling, and the REACTER protocol to improve accessibility for particle-based modeling in materials science and computational chemistry.
Key Contributions
- The paper introduces an eight-tutorial suite that guides users from basic simulation setups to advanced techniques including reactive force fields, grand canonical Monte Carlo, enhanced sampling, and the REACTER protocol. These step-by-step workflows provide standardized procedures for complex physics selections and parameter configurations in LAMMPS.
- The work presents LAMMPS-GUI, a cross-platform graphical text editor that integrates input editing, direct execution, data extraction, and system visualization for LAMMPS workflows. This interface serves as the primary tool across all tutorials to manage simulation inputs, execution, and analysis.
- Concrete implementation examples demonstrate free energy profile calculations using free sampling and umbrella sampling methods. These documented procedures provide adaptable templates for broader applications, including adsorption barrier analysis and membrane translocation studies.
Introduction
Molecular simulations are foundational to materials science and computational chemistry, enabling researchers to predict structural, thermodynamic, and dynamic properties that bridge computational models with experimental data. While open-source platforms like LAMMPS deliver the parallel computing power needed to model complex atomic and coarse-grained systems, their steep learning curve and dense documentation frequently overwhelm newcomers. Navigating intricate input scripting, selecting appropriate force fields, and configuring thermodynamic ensembles often stall progress, effectively creating a black box that limits broader scientific adoption. To resolve these bottlenecks, the authors introduce a structured suite of eight tutorials that guide users from fundamental simulations to advanced techniques like reactive force fields and enhanced sampling. They also release LAMMPS-GUI, a cross-platform graphical editor that streamlines input creation, execution, and data visualization. This combined educational and software framework flattens the entry barrier, empowering researchers to reliably design and interpret molecular dynamics and Monte Carlo workflows.
Dataset
Dataset Composition and Sources
- The authors provide a collection of eight educational tutorials hosted on GitHub and accessible through LAMMPS-GUI.
- The resources require LAMMPS stable release v22Jul2025 and LAMMPS-GUI v1.7.0, with support for Linux, macOS, and Windows.
- External tools such as VMD, OVITO, Python libraries, and plotting software are referenced for visualization and analysis.
Key Details for Each Subset
- Tutorial 1: Binary Lennard-Jones fluid system containing 1500 atoms of type 1 and 100 atoms of type 2. Simulations perform energy minimization and run in NVE and NVT ensembles.
- Tutorial 2: Carbon nanotube model utilizing OPLS-AA and AIREBO force fields. The system undergoes external deformation to demonstrate bond breaking and visualization.
- Tutorial 3: Solvated polymer system combining flexible SPC/Fw water molecules and a polymer chain. The setup includes 700 water molecules and uses NPT ensemble equilibration with long-range electrostatic solvers.
- Tutorial 4: Confined electrolyte between walls using the rigid TIP4P/2005 water model. The tutorial demonstrates non-equilibrium MD with shear flow.
- Tutorial 5: Reactive system utilizing the ReaxFF force field for dynamic chemical reactions and charge equilibration.
- Tutorial 6: Fluid adsorption in silica pores modeled via grand canonical Monte Carlo simulations to simulate open systems exchanging particles with a reservoir.
- Tutorial 7: Advanced free energy method using umbrella sampling to calculate energy barriers for difficult-to-sample landscapes.
- Tutorial 8: Nylon-6,6 polymerization simulation with a carbon nanotube embedded in the melt. The REACTER protocol tracks polymerization and water formation over time.
Data Usage and Processing
- The authors use this collection as a learning resource rather than a training dataset for machine learning models.
- Users follow the tutorials sequentially to generate simulation input files and analyze results. There are no training splits, mixture ratios, or cropping strategies.
- Processing involves defining regions, creating atoms with random placement and overlap constraints, and importing topologies via data files.
- Metadata is constructed within input files and data files specifying atom types, force field parameters, and bond definitions.
- Trajectory visualization is handled using dump commands to generate image sequences, with options to adjust rendering styles and camera views.
Method
The authors leverage LAMMPS-GUI, a graphical text editor designed for creating and executing LAMMPS input scripts, to streamline the setup and execution of molecular dynamics simulations. The core workflow involves writing a structured input script that defines the simulation's global parameters, system, and dynamics. This script is organized into distinct, logical categories: Initialization, System definition, Settings, Monitoring, and Run. The Initialization section establishes fundamental properties such as the unit system (e.g., units lj), dimensionality (e.g., dimension 3), atom style (e.g., atom_style atomic), and boundary conditions (e.g., boundary p p p for periodic boundaries in all directions). The System definition section proceeds to create the simulation box (e.g., create_box) and populate it with atoms (e.g., create_atoms). The Settings section specifies the interatomic potential (e.g., pair_style lj/cut 4.0), assigns masses to atom types (e.g., mass), and defines the Lennard-Jones parameters (e.g., pair_coeff) for interactions between different atom types. The Monitoring section configures the output of thermodynamic data (e.g., thermo_style custom) and visualization (e.g., dump image). The Run section contains the commands that drive the simulation, such as minimize for energy minimization and fix nve or fix nvt for molecular dynamics, with the run command initiating the simulation steps. 
The simulation framework is built upon the LAMMPS molecular dynamics engine, which executes the commands in the input script sequentially from top to bottom. The system's energy and forces are calculated based on the specified interatomic potential, which is defined by the pair_style and pair_coeff commands. For a Lennard-Jones potential, the energy between two particles is given by Eij(r)=4ϵij[(rσij)12−(rσij)6], where r is the inter-particle distance, ϵij is the interaction strength, and σij is the distance at which the potential energy is zero. LAMMPS supports various force fields, including many-body potentials like ReaxFF and AIREBO, which are used to model bond breaking and formation. The simulation's statistical ensemble is controlled by the fix commands; for instance, fix nve integrates Newton's equations of motion using the velocity-Verlet algorithm, resulting in a microcanonical (NVE) ensemble, while fix nvt applies a Nosé-Hoover thermostat to maintain a constant temperature, resulting in a canonical (NVT) ensemble. 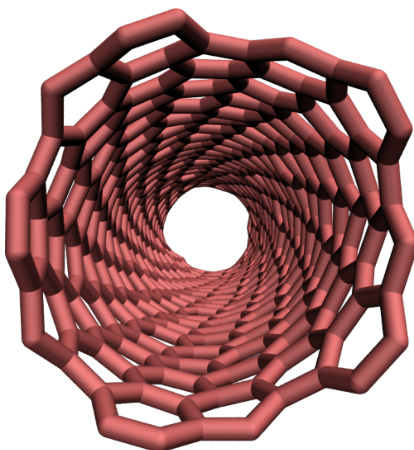
To analyze the simulation results, LAMMPS provides a suite of tools. The thermo command outputs thermodynamic properties (e.g., temperature, pressure, energy) at specified intervals, and the thermo_style command defines the format of this output. The dump command writes out the system's state, including atomic positions, velocities, and other data, to a file for visualization or post-processing. Visualization is facilitated by the dump image command, which generates images of the system at specified time steps. For more advanced analysis, LAMMPS allows the use of compute commands to calculate quantities like the radius of gyration or coordination number, which can then be used in thermo_style or dump commands. The fix commands, such as fix gcmc for grand canonical Monte Carlo or fix bond/react for reactive simulations, can be used to perform specialized sampling or to model chemical reactions. The results are often exported in formats like YAML or CSV for analysis in external software like Python. 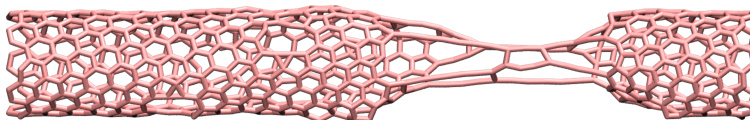
The framework also supports advanced simulation techniques. For example, the fix deform command can be used to apply a constant strain rate to the simulation box, allowing the study of material deformation. The fix addforce command can apply a constant external force to specific groups of atoms, enabling the simulation of processes like pulling a polymer. For calculating free energy profiles, the fix gcmc command can be used for grand canonical Monte Carlo simulations, and the fix spring command can be used in umbrella sampling to bias the system along a reaction coordinate. The results from these biased simulations are then analyzed using algorithms like the Weighted Histogram Analysis Method (WHAM) to reconstruct the unbiased free energy profile. 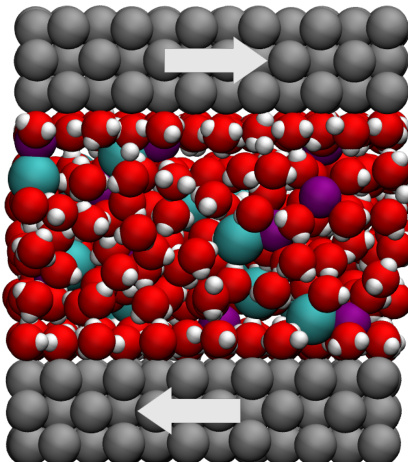
Finally, the framework supports the use of external visualization tools. The dump custom command can be used to write out a trajectory file that can be imported into software like VMD or OVITO. The rerun command allows for the analysis of a simulation's trajectory without having to re-run the simulation, enabling the computation of properties not initially set up. The write_data and write_restart commands allow for saving the system's state to a file, which can be used to restart a simulation from a previous point or to transfer data between different simulations. The read_data command reads this saved state back into LAMMPS, allowing for the continuation of a simulation or the modification of an existing system. 
Experiment
Molecular dynamics simulations using LAMMPS evaluate atomic relaxation, mixing dynamics, mechanical deformation, and thermodynamic consistency across distinct systems. Energy minimization and Langevin-driven mixing experiments validate that disordered initial configurations naturally evolve toward stable, lower-energy states characterized by increased cross-species coordination. Tensile deformation tests comparing conventional and reactive force fields demonstrate the accurate capture of elastic stretching followed by bond rupture and plastic failure. Additionally, free sampling and polymerization runs confirm that the computational framework reliably reproduces imposed thermodynamic potentials and maintains stable reaction kinetics during chain formation.
The authors perform an energy minimization simulation to reduce the potential energy of a system from a positive initial value to a negative final value, indicating atoms moving to more stable configurations. The simulation tracks the potential energy over time, showing a rapid initial decrease followed by a gradual approach to a plateau, with the total energy remaining constant as kinetic energy is zero throughout the minimization process. The potential energy decreases from a positive value to a negative value during minimization. The energy reduction is rapid initially and then slows, plateauing at a stable negative value. The total energy remains constant as the kinetic energy is zero throughout the minimization process.
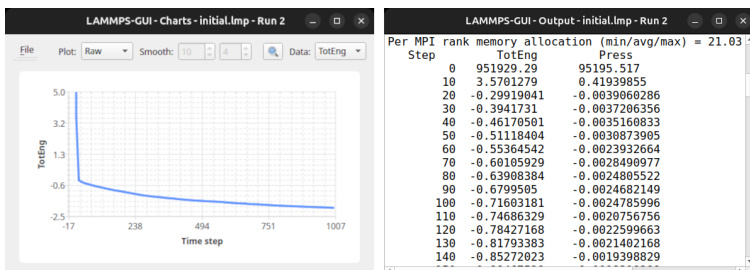
The experiment employs an energy minimization simulation to evaluate atomic stabilization over time. By tracking potential energy throughout the process, the results demonstrate a rapid initial decline followed by a gradual approach to a stable negative plateau. This qualitative trend confirms that the system successfully transitions to lower energy configurations while maintaining constant total energy due to zero kinetic energy. Overall, the simulation validates the effectiveness of the minimization approach in achieving structural stability.