Command Palette
Search for a command to run...
VASP على معالج الرسوميات: تطبيق على حسابات التبادل الدقيق لاستقرار البورون العنصري
VASP على معالج الرسوميات: تطبيق على حسابات التبادل الدقيق لاستقرار البورون العنصري
M. Hutchinson M. Widom
VASP مع Phonopy: حساب تشتت الفونونات للسيليكون
الملخص
توفر وحدات معالجة الرسومات ذات الأغراض العامة (GPUs) سرعات معالجة عالية لفئات معينة من الحسابات القابلة للتوازي العالي، مثل عمليات المصفوفات وتحويلات فورييه، والتي تشكل جوهر حسابات البنية الإلكترونية القائمة على المبادئ الأولى. يؤدي تضمين التبادل الدقيق إلى زيادة تكلفة نظرية الدالة الكثافية بعوامل كبيرة، مما يحفز استخدام وحدات معالجة الرسومات. يؤدي نقل كود كثافة الإلكترونات الواسع الانتشار VASP ليعمل على وحدة معالجة الرسومات إلى تحقيق دفعة أداء تتراوح بين 5 إلى 20 ضعفاً للتبادل الدقيق مقارنةً بمعالج وحدة المعالجة المركزية التقليدي. نقوم بتحليل اختناقات الأداء ونناقش فئات المشكلات التي ستستفيد من وحدات معالجة الرسومات. وكأمثلة على قدرات هذا التنفيذ، نحسب استقرار الشبكات البلورية لبنيتي البورون سداسية الأوجه ألفا وبيتا باستخدام التبادل الدقيق. تؤكد نتائجنا الأفضلية الطاقية للاحتلال الجزئي لكسر التماثل في بنية بيتا سداسية الأوجه عند درجات الحرارة المنخفضة، إلا أنها لا تحسم مسألة الاستقرار النسبي بين ألفا وبيتا.
One-sentence Summary
Porting the VASP density functional theory code to GPU architectures yields a 5- to 20-fold speedup over CPU implementations for exact-exchange calculations, as demonstrated by low-temperature lattice stability evaluations of elemental boron that confirm a symmetry-breaking partial occupation in the beta-rhombohedral phase without resolving its stability relative to the alpha structure.
Key Contributions
- The paper presents a GPU-accelerated port of the VASP electronic structure code that efficiently computes exact-exchange terms, which traditionally increase density functional theory costs by orders of magnitude. This implementation exploits the high parallelism of matrix operations and Fourier transforms to achieve a 5- to 20-fold performance increase over conventional CPU execution.
- Computational bottlenecks within the exact-exchange routines are analyzed to identify specific electronic structure problem classes that benefit most from GPU acceleration. This evaluation establishes optimization strategies for scaling parallelized quantum chemistry workflows.
- The accelerated framework calculates the relative lattice stability of alpha and beta-rhombohedral boron polymorphs using the HSE06 hybrid functional. Benchmark results confirm an energetic preference for the symmetry-breaking partial occupation of the beta-rhombohedral structure at low temperatures.
Introduction
First-principles quantum mechanical simulations are essential for predicting material properties but face severe computational bottlenecks as system size increases. Determining the stable crystal structure of complex elements like boron requires exceptionally accurate electronic structure methods, yet standard density functional theory approximations cannot resolve the minute energy differences between competing polymorphs. Higher-accuracy hybrid functionals that include exact electron exchange are computationally prohibitive on traditional processors, historically limiting their use to smaller systems. The authors leverage GPU acceleration to port the VASP codebase, specifically optimizing exact-exchange calculations for massive parallelism. This approach delivers up to twenty-fold performance gains over CPUs while enabling precise energy comparisons that resolve long-standing questions about boron structural stability.
Dataset
- Dataset composition and sources: The authors compile a benchmark set of four crystalline boron phases generated through first-principles density functional theory (DFT) calculations using the VASP software package.
- Subset details: The collection includes a 12-atom α-rhombohedral structure (hR12), a 96-atom supercell of α (hR12x8) constructed by doubling the lattice axes to match β parameters, a 105-atom ideal β-rhombohedral structure (hR105), and a 107-atom symmetry-broken β variant (aP107) optimized for GGA total energy.
- Data usage and processing: The authors use these structures to benchmark computational efficiency and energy convergence between conventional DFT and hybrid HF-DFT methods. They prioritize 2x2x2 k-point meshes for efficient benchmarking while reserving 3x3x3 meshes for high-precision convergence tests to resolve energy differences at the meV/atom level.
- Mesh and grid configuration: Monkhorst-Pack k-point meshes are adjusted for structural symmetry and lattice scaling, with real-space FFT grids scaling proportionally with lattice dimensions. All runs employ Projector Augmented Wave potentials, the PBE exchange-correlation functional, a fixed 319 eV plane wave cutoff, and PREC=Normal settings to ensure accurate charge density representation without wrap-around errors.
Method
The authors leverage a GPU-accelerated implementation of the VASP code to perform exact-exchange calculations for electronic structure simulations, with a focus on enhancing computational performance for demanding tasks such as those involving elemental boron. The framework is designed to integrate GPU acceleration into the existing VASP architecture while preserving its modular and abstraction-based structure. The implementation involves intercepting the main execution flow at five key points: initialization and cleanup routines, the FFT3D routine for large-scale transforms, and the FOCK_ACC and FOCK_FORCE routines. These intercepts allow selective offloading of computationally intensive operations to the GPU, while smaller or less intensive tasks remain on the CPU.
The core computational task in exact-exchange calculations involves a four-nested loop structure: the outer two loops iterate over k-point indices, and the inner two over band indices, computing contributions to the Fock exchange energy. The authors optimize this by offloading the inner loops—specifically the band-related operations—to the GPU. This strategy transforms matrix-vector operations into more efficient matrix-matrix operations, enabling higher parallelization and better utilization of GPU resources. The memory footprint on the GPU is dynamically controlled through the NSIM parameter, which manages the number of bands processed concurrently. To minimize data transfer overhead between the host and device, non-intensive operations within the loops are also executed on the GPU.
The implementation relies on the cuFFT and cuBLAS libraries for high-performance FFT and BLAS operations, respectively. Additionally, 20 custom CUDA kernels were developed to replicate the CPU functionality, ensuring compatibility with VASP's existing computational logic. Asynchronous execution is achieved through CUDA streams, enabling concurrent execution of smaller kernels and improving performance, particularly for smaller input structures. The framework supports both full double precision and mixed precision arithmetic, with numerical differences between the two settings found to be negligible—within one thousandth of a percent—demonstrating robust accuracy.

Experiment
The study evaluates the computational performance and physical accuracy of GPU-accelerated exact-exchange density functional theory by benchmarking a dual-GPU workstation against multi-core CPU supercomputer configurations across various boron crystal structures. The performance experiments validate that GPU acceleration effectively circumvents CPU communication bottlenecks, delivering execution times that rival large-scale cluster runs and making previously impractical hybrid functional calculations feasible on desktop hardware. Concurrently, the structural energetics analysis demonstrates that exact-exchange methods favor alpha-boron over certain beta-polymorphs, with energy convergence patterns across functionals indicating that higher-level theoretical approaches may still be necessary to definitively resolve boron's stable low-temperature phase. Collectively, these results establish GPU-based acceleration as a scalable, cost-effective strategy to expand the application of exact-exchange simulations to larger materials systems and dynamic processes.
The authors analyze the performance of GPU-accelerated calculations for boron structures, comparing runtime components across different configurations and platforms. Results show significant speedups for GPU implementations in specific computational routines, with performance scaling favorably against CPU-based systems, especially for smaller structures where communication overhead is limited. GPU acceleration provides substantial speedups in key computational routines compared to CPU-only implementations. Performance gains are most pronounced for smaller structures due to reduced communication overhead on GPU systems. The GPU system achieves comparable performance to multi-core CPU setups, enabling high-level calculations on desktop hardware.
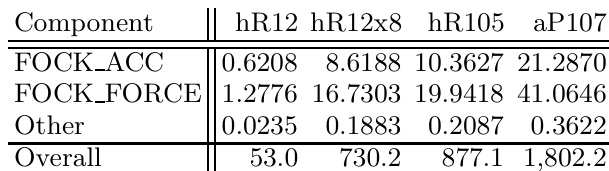
The authors compare the performance of GPU and CPU systems for calculating structural energies of boron structures using VASP with hybrid functionals. Results show that the GPU implementation significantly outperforms single CPU cores, enabling calculations that would otherwise require large supercomputing resources to be performed on desktop hardware. The performance advantage of GPUs is particularly pronounced for larger structures, where the GPU system achieves comparable or better times-to-solution than many-core CPU configurations. GPU acceleration enables exact-exchange calculations on desktop hardware that would otherwise require supercomputing resources. The GPU system achieves comparable performance to many CPU cores on the supercomputer for large structures. The performance advantage of GPUs increases with structure size, making them more effective for larger systems.
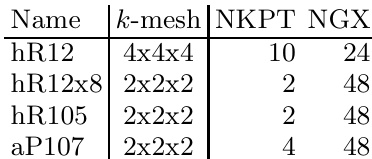
The authors present a comparison of structural energies calculated using different density functionals, showing significant differences between PBE and HF methods. The results indicate that the HF method yields higher energy values for the same structures, suggesting a notable impact of the exchange-correlation functional on the predicted stability of boron structures. The HF method predicts higher structural energies compared to the PBE method for all tested configurations. The energy differences between PBE and HF are more pronounced for certain k-meshes, indicating sensitivity to computational parameters. The comparison highlights the importance of the choice of functional in determining the relative stability of boron structures.
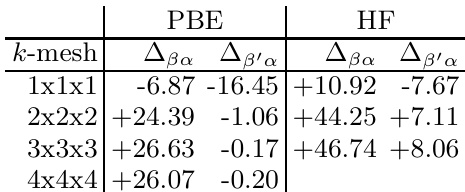
The authors compare structural energies across different density functionals, showing that the energy of the beta structure is higher than that of the alpha structure, and the optimized variant of the hR141 structure reduces the energy per atom. The results indicate convergence toward local density approximation values, suggesting the need for higher-level theory to resolve the stability of boron structures. The beta structure has higher energy compared to the alpha structure across all functionals. The optimized variant of the hR141 structure shows lower energy per atom than the ideal beta structure. Energy values converge toward local density approximation rather than generalized gradient approximation.
The authors compare the performance of CPU and GPU implementations for calculating structural energies of boron structures, focusing on run-times and computational efficiency. Results show that the GPU implementation significantly outperforms single CPU cores, enabling the study of complex structures that were previously impractical on standard hardware. The GPU achieves substantial speedups in key computational routines, making high-level calculations accessible on desktop systems. The GPU implementation achieves substantial speedups over single CPU cores, enabling previously impractical calculations on desktop hardware. The GPU performs significantly faster than CPU-only systems, with up to an order of magnitude improvement in computational efficiency. The GPU's high performance allows for the study of complex structures that would otherwise require supercomputing resources.
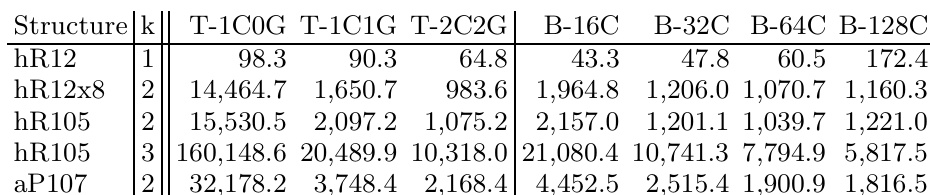
The study evaluates GPU-accelerated density functional theory calculations for boron structures, comparing hardware performance across platforms and validating the influence of exchange-correlation functionals on predicted stability. Benchmarks demonstrate that GPU implementations deliver substantial computational speedups over single-core CPUs, making high-level hybrid functional calculations feasible on desktop systems with performance advantages that scale favorably for larger structures. Functional comparisons reveal that hybrid methods consistently yield higher structural energies than gradient approximations, with results highly sensitive to computational parameters. Furthermore, cross-functional analysis of specific boron polymorphs indicates that the beta phase is less stable than the alpha phase, while optimized structures converge toward local density approximation limits, underscoring the need for advanced theoretical methods to definitively resolve boron phase stability.