Command Palette
Search for a command to run...
100여 개 대학이 세계 최대 규모의 다중 코호트 단백질유전체학 연구를 시작했으며, 이를 통해 약 8만 명의 참가자로부터 얻은 데이터를 기반으로 질병을 유발하는 유전자를 밝혀내고 기존 약물의 용도를 변경할 수 있을 것으로 기대된다.

인간의 게놈은 마치 삶에 대한 완벽한 설명서와 같아서 외모, 키, 체격, 질병 위험도 등 모든 유전 정보를 기록하고 있습니다. 하지만 이 설명서를 해독하는 것은 간단한 과정이 아닙니다. 특정 질병에 걸리기 쉽게 만드는 병원성 돌연변이를 비롯한 다양한 "예상치 못한 사건"이 발생할 수 있기 때문입니다. 더욱 어려운 것은...대부분의 병원성 변이는 단백질을 직접 코딩하지 않는 유전체의 비코딩 영역에 위치합니다.질병을 유발하는 유전자가 무엇인지, 또는 어떤 기전을 통해 질병을 일으키는지 명시하지 않는 이러한 "블랙박스" 메커니즘은 병원성 유전자와 기전을 추론하는 우리의 능력을 심각하게 제한합니다. 그리고 유전자 기능을 실제로 구현하는 직접적인 실행자로서,인간의 혈액 속에 순환하는 수천 가지 단백질은 블랙박스 메커니즘을 해독하고 비코딩 변이와 질병 관련 메커니즘을 연결하는 데 핵심적인 역할을 합니다.
현재 프로테오게놈학 연구는 임상 병인 및 잠재적 약물 표적 분야에서 상당한 진전을 이루었지만, 인간 생물학에 체계적이고 대규모로 적용하는 데에는 여전히 한계가 있습니다. 첫째, 과거 연구는 거의 전적으로 근접한 시스 작용 변이체(즉, 시스 단백질 양적 형질 유전자좌, cis-pQTL)에 초점을 맞추었습니다.비코딩 변이는 조절 영역에 위치할 수 있으며, 따라서 인접한 여러 코딩 유전자에 직접적인 영향을 미칠 수 있습니다.또한, 유전자는 유전체 내 다른 위치에 있는 유전자가 코딩하는 단백질을 원거리에서 간접적으로 조절할 수 있습니다. 둘째, 질병 진단 및 예후에 영향을 미치는 단백질 바이오마커의 다중 유전자 유전 구조에 대한 기존 연구는 아직 불충분합니다. 마지막으로, 단백질 양적 형질 유전자좌를 안정적이고 보편적으로 식별하기 위해서는 다양한 인구 집단에서 반복적인 검증이 필요합니다.현재 광범위 단백질체학 분야에서 이러한 종류의 인체 검증 연구는 매우 드물게 진행되고 있습니다.
이를 고려하여,퀸 메리 런던 대학교와 케임브리지 대학교를 포함한 100여 개 대학 및 연구 기관의 연구팀이 현재까지 세계 최대 규모의 다중 코호트 단백질유전체학 연구 결과를 발표했습니다.38개의 독립적인 연구 코호트와 총 78,664명의 피험자를 대상으로 한 대규모 단백질-혈당 유전체 메타분석을 기반으로, 24,738개의 단백질 정량적 형질 부위가 체계적으로 식별되었고 1,116개의 순환 단백질과 연관되어 단백질 수준에서 광범위한 근접 및 거리 유전적 조절 특성을 종합적으로 밝혀냈다.
머신러닝을 활용하여 순환 단백질의 양을 조절하는 주요 경로, 세포 유형 및 조직 기원을 추가적으로 분석하고, 단백질 조절 네트워크에서 N-글리코실화의 핵심적인 역할을 규명했습니다. 나아가, 단백질의 시스(cis) 및 트랜스(trans) 조절 차이를 구분함으로써 다양한 생물학적 표현형의 내재적 메커니즘을 효과적으로 밝혀내고, 특정 질병에 대한 잠재적인 단백질 약물 표적을 선별하는 데 필요한 근거를 제공했습니다. 또한, 트랜스 부위에 대한 삼각 분석을 통해 "약물 재활용(drug repurposing)"에 대한 더욱 심층적인 근거를 발견했습니다.
"다중 코호트 프로테오게놈 분석을 통해 프로테옴 및 질병체 전반에 걸친 유전적 효과를 밝혀냈다"라는 제목의 관련 연구 결과는 Cell 저널에 게재되었습니다.
연구 하이라이트:
* 현재까지 진행된 가장 큰 규모의 다중 코호트 단백질유전체학 연구로, 38개의 독립적인 연구 코호트와 총 78,664명의 참가자를 포함합니다.
* 24,738개의 단백질 양적 형질 유전자좌를 식별하고 이를 1,116개의 순환 단백질과 연관시켜 단백질 수준에서 광범위한 근접 및 장거리 유전적 조절 특성을 종합적으로 밝혀냈습니다.
* 본 연구는 순환 단백질의 유전적 수준에서의 조절 메커니즘을 체계적으로 규명하여, 인간 질병의 분자 메커니즘을 이해하고, 혁신적인 치료 표적을 발굴하며, 약물 재활용 연구를 수행하는 데 중요한 이론적 기반과 데이터 자원을 제공합니다.

서류 주소:
https://www.cell.com/cell/fulltext/S0092-8674(26)00385-5
최대 규모 핵심 데이터: 38개 국제 코호트, 약 8만 명의 참가자
본 연구는 세계 최대 규모의 다중 코호트 단백질유전체 메타분석을 나타냅니다.유럽계 참가자 78,664명을 대상으로 한 38개의 국제 코호트를 통합하여,Olink 고처리량 프로테오믹스 기술을 사용하여 1,161개의 혈액 단백질 표적을 분석한 결과, 24,738개의 정밀하게 매핑된 pQTL(5,040개의 cis-pQTL 및 19,698개의 trans-pQTL 포함)이 확인되었고, 1,116개의 유효 단백질에 대한 유전적 조절 데이터가 얻어졌다.
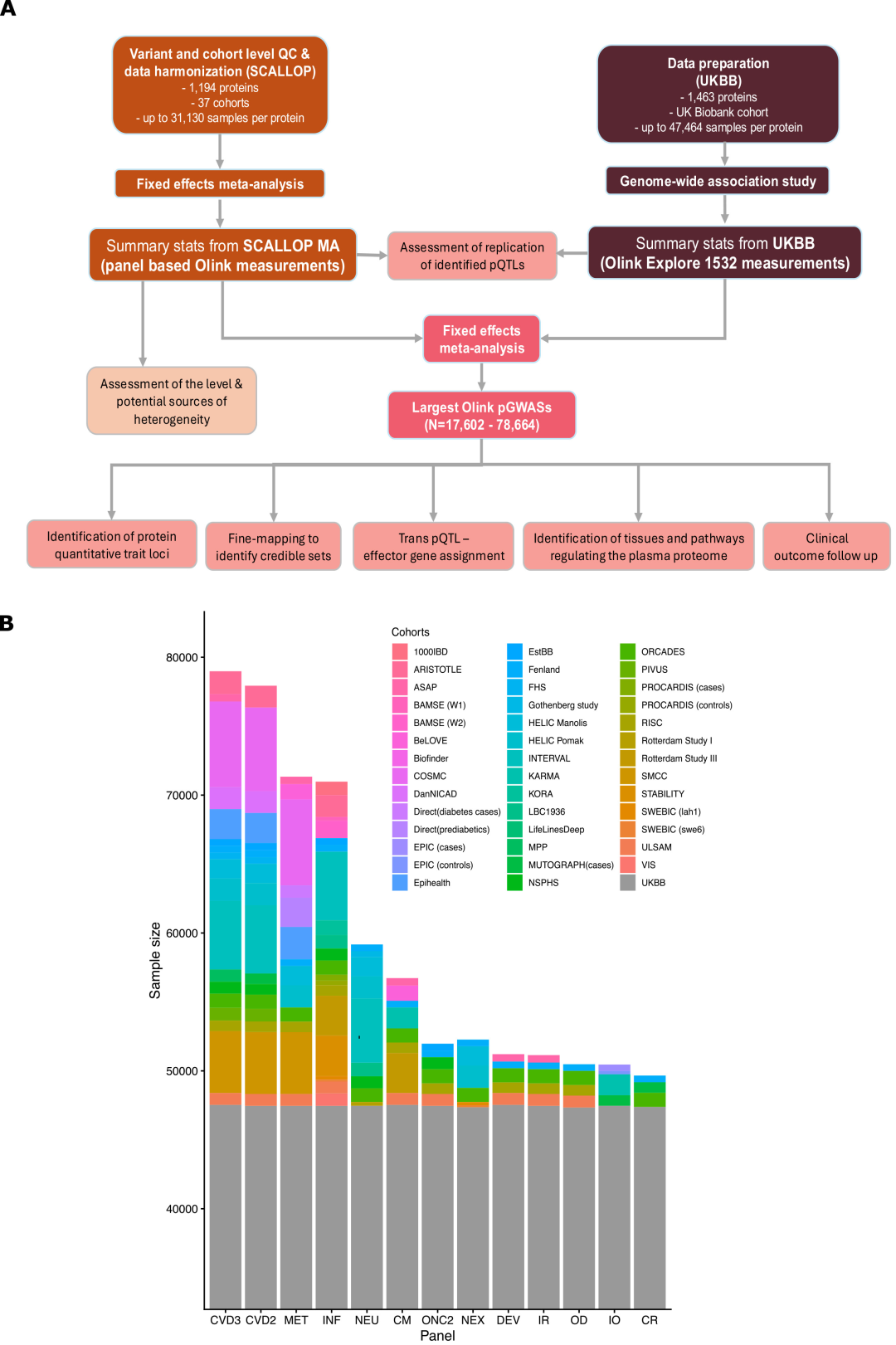
SCALLOP 메타 분석:이 데이터에는 37개 코호트의 전체 게놈 통계와 1,194개의 혈액 단백질 표적이 포함되어 있으며, 참가자의 대다수는 유럽계입니다. 항체 기반 단백질체학 분석은 Olink에서 제공하는 13개의 Target-96 분석 패널 중 하나 이상을 사용하여 수행되었으며, 각 패널은 심혈관, 면역, 염증, 신경 및 대사 분야를 포괄하는 92개의 단백질 표적을 검출할 수 있습니다.
영국 바이오뱅크(UKBB):본 연구에는 유럽계 참가자 48,017명이 포함되었습니다. 연구에 사용된 단백질체 측정 데이터는 Olink Explore 1536 플랫폼을 통해 생성되었으며, 항체 기반 기술을 이용하여 1,463개의 단백질 표적을 측정했습니다.
단계별 머신러닝 분류기
본 연구의 핵심 목표는 머신러닝 모델을 활용하여 주요 조직적합성복합체(MHC) 영역 외부에 위치한 모든 trans-pQTL에 "효과 유전자"를 체계적이고 정확하며 대규모로 할당하는 것입니다. 이는 혈중 단백질 수치와 관련된 단백질 양적 형질 유전자좌(PTL)의 멀리 떨어진 유전체 영역에서 효과 유전자를 찾는 오랜 난제를 해결하는 것입니다. 이를 위해,ProGeM 아키텍처에서 영감을 받아 연구원들은 단계별 머신러닝 분류기를 개발했습니다.
첫째, 특징 및 주석의 출처와 관련하여 연구진은 각 유전 변이 또는 그 대체 변이에 대해 다차원적인 생물학적 및 유전체 주석을 통합했습니다(r² > 0.6). 변이 수준 주석에는 1Mb 염기 창 내에서 변이와 유전체 사이의 거리 및 변이 효과 예측(VEP) 도구를 기반으로 추론된 잠재적 기능적 영향이 포함되었습니다.
동시에, 1Mb 염기서열 창 내의 각 유전자에 대해 유전자 수준의 주석 작업을 수행했습니다. 여기에는 GTEx v8 단백질 풍부도-유전자 발현을 기반으로 한 QTL 공동 위치 관련 증거 확보, 희귀 변이 부하 연관성 분석, OmnipathR 버전 3.10.1 패키지를 사용한 문헌 검토를 통해 전이 유전자에 의해 코딩된 시스 단백질에 해당하는 리간드-수용체/단백질 복합체의 존재 여부 확인, 그리고 KEGG/REACTOME 주석 정보를 기반으로 관련 유전자가 동일한 생물학적 경로에 참여하는지 여부 확인 등이 포함되었습니다.
다음으로, 머신러닝 모델에 필요한 훈련 데이터셋을 구축하는 단계로 넘어가지만, 유전자 할당에 널리 사용되는 표준 변이체가 부족하기 때문에,연구진은 생물학과 유전체학에 대한 사전 지식을 활용하여 부분적으로 독립적인 세 가지 "추정되는 참양성(PTP)" 세트를 얻었습니다.편향을 방지하기 위해 각 PTP 세트 내에서 하나의 시스 단백질만 유지하고 1Mb 창 내의 다른 유전자들은 음성 샘플로 간주했습니다. 구체적으로, 여기에는 리간드-수용체 쌍을 코딩하거나 시스 단백질과 높은 신뢰도의 단백질 복합체를 형성하는 트랜스 유전자(n = 540), 기능적 변이에 매핑된 센티널 트랜스-pQTL(n = 1747), 그리고 유의미한 희귀 변이 부하를 갖는 트랜스 유전자(n = 1049)가 포함되었습니다. 그런 다음 훈련 세트와 테스트 세트를 유전체 영역을 기준으로 7:3 비율로 나누고, 안정성을 확보하기 위해 결과를 10회 반복했습니다.
또한, 모델 아키텍처 및 학습 과정과 관련하여 본 연구에서는 랜덤 포레스트 분류기를 모델 알고리즘으로 채택하였다. 10개의 훈련 데이터셋을 입력으로 사용하여 3겹 교차 검증을 반복적으로 수행하고, 서브샘플링 전략을 결합하여 학습 중 데이터셋 불균형 문제를 해결하였다.모델 학습은 R 언어 caret v6.0.94 툴킷을 사용하여 수행되었으며, 각 학습 세트에서 가장 성능이 우수한 랜덤 포레스트 모델은 카파 점수 평가를 통해 선정되었습니다.
그런 다음, 각 가상의 참양성 데이터 세트에 해당하는 10개의 랜덤 포레스트 분류기를 사용하여 trans-pQTL의 모든 후보 효과 유전자에 대해 하나씩 점수를 매겼습니다. 동일한 가상의 참양성 데이터 세트에 대한 10개 분류기의 점수 중 중앙값을 먼저 구한 다음, 세 가지 예측 점수를 합산했습니다. 동시에, 각 가상의 참양성 데이터 세트에 대한 분류 모델을 구축할 때, 참양성 샘플을 정의하는 데 사용된 특징 변수들을 제거했습니다.
결론적으로, 세 가지 분류 모델 모두 0.54에서 0.57 사이의 중간 카파 계수를 나타내며 안정적이고 신뢰할 수 있는 성능을 보였다.
병인 기전을 해독하는 것은 신약 개발 및 기존 약물의 용도 변경을 위한 유전적 근거를 제공합니다.
본 연구는 38개의 국제 코호트와 78,664명의 참가자를 기반으로 1,161개의 혈액 단백질을 표적으로 하는 다중 코호트 단백질 유전체 메타분석을 수행하여 순환 단백질 수치의 유전적 조절 패턴과 질병과의 연관성을 체계적으로 규명했습니다.
pQTL 식별 및 특성
본 연구에서는 14,690개의 지역적 센티널 변이를 확인했으며, 베이지안 정밀 매핑을 통해 5,040개의 cis-pQTL과 19,698개의 trans-pQTL을 포함하는 24,738개의 독립적이고 신뢰할 수 있는 변이 세트를 도출했습니다. 이 중 cis-pQTL은 87.1% 위치의 단백질에서, trans-pQTL은 94.1% 위치의 단백질에서 발견되었습니다. 또한, 82.3% 위치의 cis-pQTL과 83.3% 위치의 trans-pQTL은 높은 신뢰도를 보이는 부위로, 278개의 새로운 cis-pQTL과 4,013개의 trans-pQTL을 포함하고 있습니다. 더욱이, 비유럽계 코호트에서 확인된 부위들의 효과 크기는 유럽계 코호트와 중간 정도의 상관관계(r = 0.6)를 보였습니다.이는 결과의 집단 간 견고성을 검증합니다.
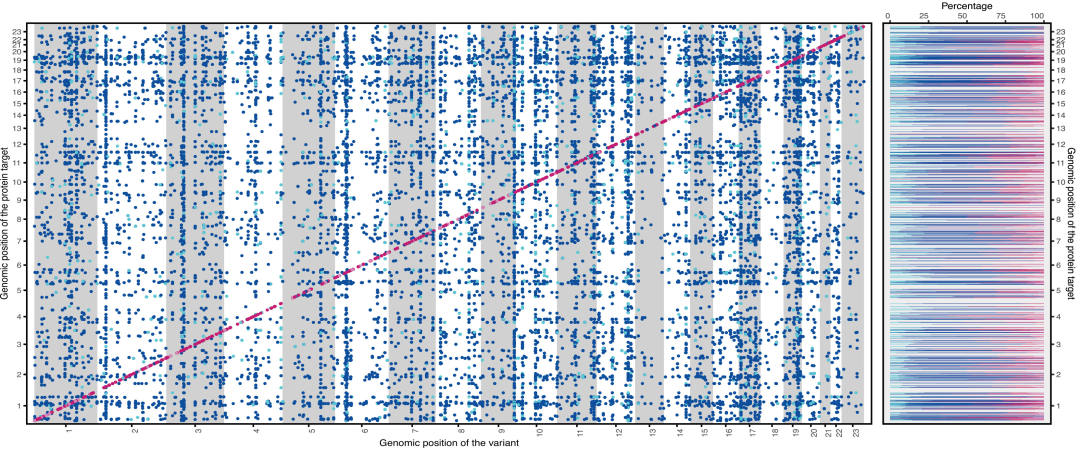
SCALLOP 및 UKBB 메타 분석에서 정밀하게 위치한 단백질 양적 형질 유전자좌
또한, 혈중 단백질 수치 변이에 대한 유전적 위치의 설명력에는 상당한 차이가 있습니다. cis-pQTL은 단백질 변이의 평균 8.41 TP3T를 설명하며, 이는 trans-pQTL보다 유의미하게 높습니다. 그러나 ICAM2 및 FUCA1과 같은 단백질은 주로 trans-pQTL에 의해 조절되며, 그 설명력은 각각 52.71 TP3T 및 68.41 TP3T인 반면, cis-pQTL은 각각 0.31 TP3T 및 6.31 TP3T만을 설명합니다.
또한, 261개의 단백질 표적에 대한 추가 관찰 결과, pQTL 변이의 설명력과 다유전자 유전율 사이에 유의미한 선형 상관관계가 없는 것으로 나타났습니다. 이는 본 연구가 이러한 단백질에 대한 pQTL 식별에 있어 거의 포화 상태에 도달했음을 시사합니다.
유전자 조절을 받는 단백질 표적의 특성
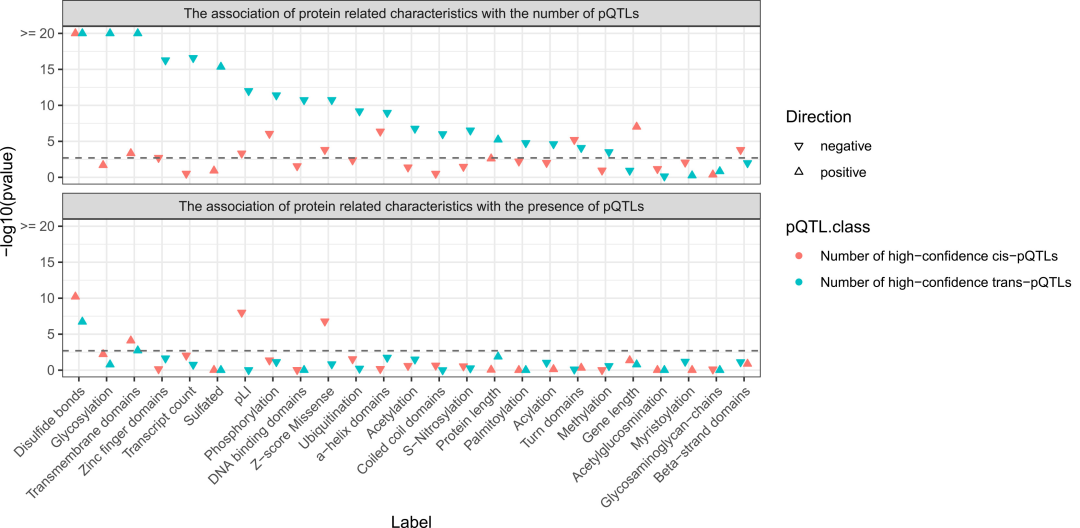
영과잉 포아송 회귀 모델을 기반으로 pQTL의 존재 및 양과 관련된 단백질 특성.
이황화 결합과 막관통 도메인을 포함하는 단백질은 pQTL이 훨씬 더 많이 존재하며, 이는 이러한 단백질이 유전적으로 더 쉽게 조절되는 이유를 설명할 수 있습니다. 반면, 단백질 코딩 유전자의 기능적 제약 강도는 cis-pQTL의 수와 유의미한 음의 상관관계를 보입니다.
높은 수의 trans-pQTL을 가진 단백질은 당화 및 황산화와 같은 분비 단백질의 특징을 유의미하게 풍부하게 나타내지만, 아연 손가락 구조 및 DNA 결합 도메인과 같은 세포 내 단백질의 특징은 결여되어 있어, 순환 단백질의 장거리 유전적 조절이 분비 경로와 밀접하게 관련되어 있음을 시사한다.
trans-pQTL 효과 유전자 및 조절 경로 분석
기존의 생물학적 지식을 기계 학습 프레임워크에 통합한 결과, 11,261개의 trans-pQTL 중 절반 이상에서 적어도 하나의 효과 유전자가 중간 정도의 신뢰도로 식별되었으며, 그중 1,534개는 높은 신뢰도로 할당되었습니다. 또한, 13,881개의 위치에서 3분의 2에 해당하는 경우, 유전자 전반에 걸친 후보 점수의 분포는 단일 원인 유전자가 가장 유력한 병원성 유전자임을 나타냈습니다.
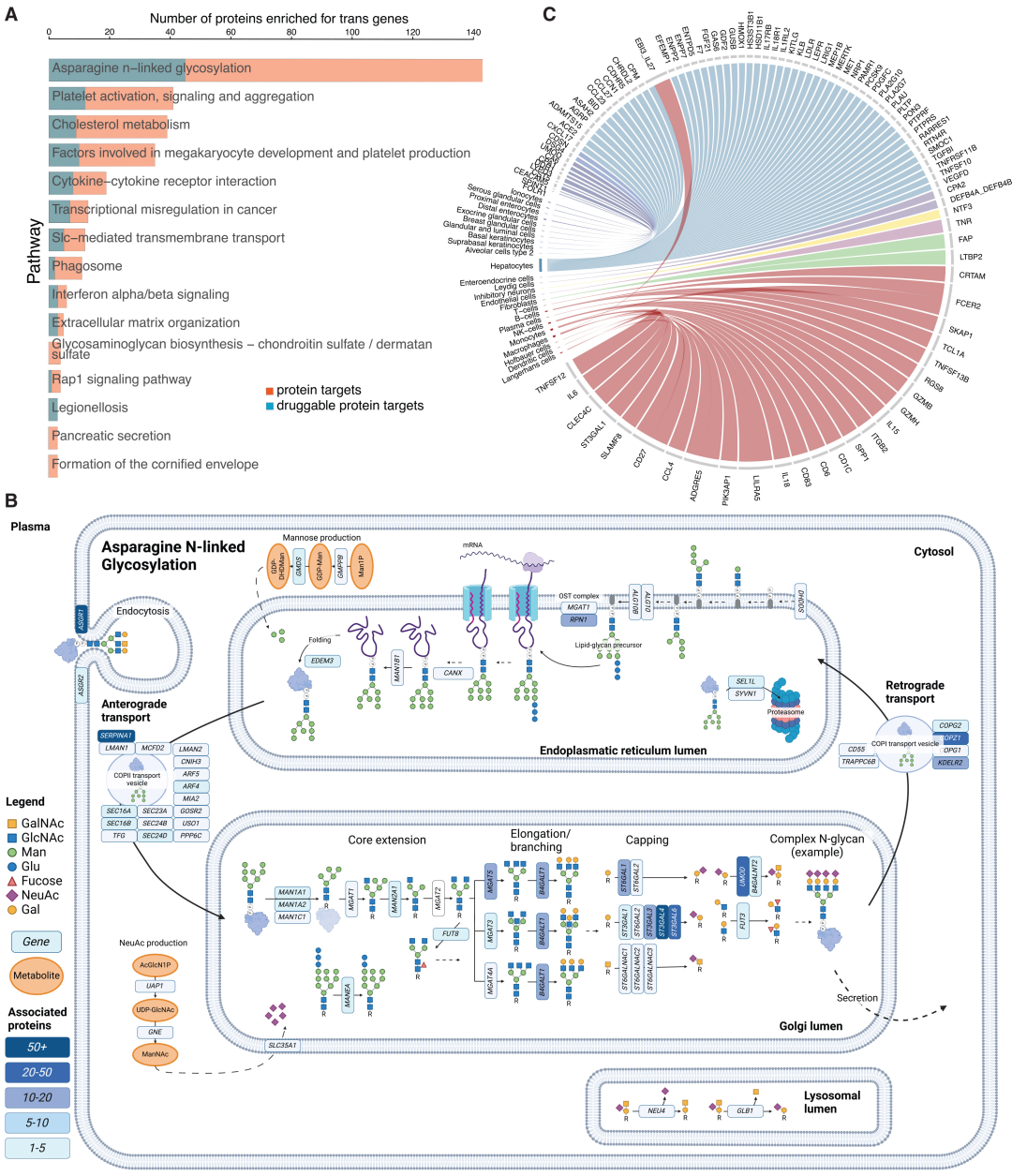
기능적 풍부도 분석 결과, 전사 효과 유전자들은 아스파라긴 N-글리코실화 경로(143개의 단백질 표적 관련)와 혈소판 활성화(41개의 단백질 표적 관련) 등에서 유의미하게 풍부하게 나타났다.N-글리코실화는 가장 흔하고 핵심적인 조절 경로입니다.
세포 및 조직 농축 분석 결과, 전사 효과 유전자는 주로 간세포, 자연 살해 세포, 내피 세포 및 제2형 폐포 세포에서 높은 발현량을 보였으며, 이는 간과 면역 세포가 순환 단백질의 원격 조절에 중요한 역할을 한다는 것을 시사합니다. 44쌍의 단백질-조직과 76쌍의 단백질-세포 유형은 비전형적인 분비 기원을 가지고 있어, 단백질 항상성 조절에 있어 장기 간 소통의 중요성을 확인시켜 줍니다.
분자 및 표현형 수준에서의 다면발현 효과
확인된 모든 독립적인 pQTL 중 43.41개의 TP3T 유전자에서 다면발현 효과가 나타났으며, 전이형 pQTL이 전이형 pQTL보다 유의미하게 높은 다면발현 효과를 보였다. 후속 연구에서는 다면발현 유전적 변이를 "분자적 다면발현", "표현형 다면발현", "비특이적 다면발현"의 세 가지 유형으로 분류했다. 이 중 절반 이상(533개 중 332개)이 표현형 다면발현 효과를 보였다.특히, 이 유전자의 발현은 간세포에서 2배 증가했으며, 단백질 복합체, 리간드-수용체 상호작용 및 경로 시너지 효과를 통해 표적 단백질을 우선적으로 조절했습니다.
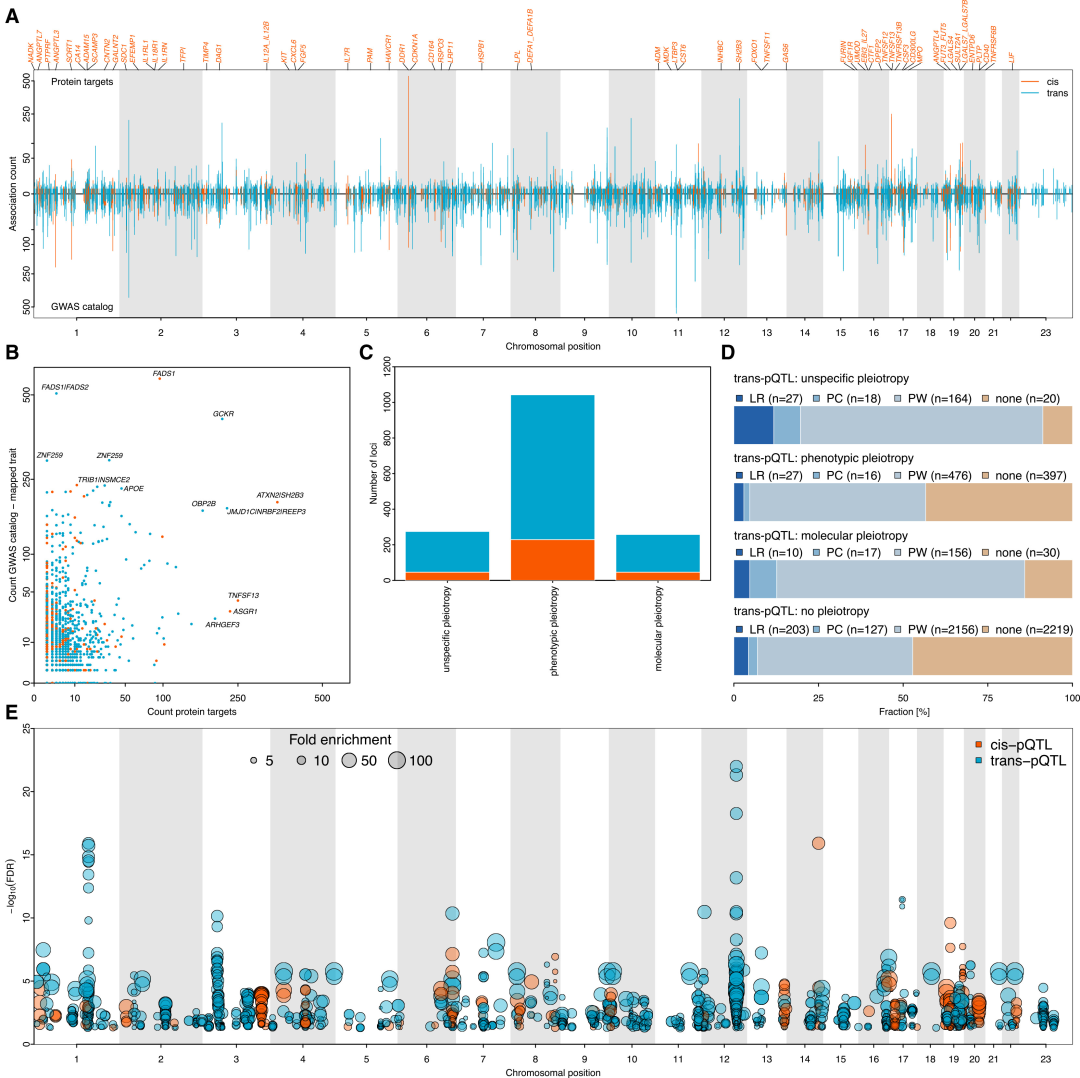
285개의 다면발현성 pQTL이 질병 GWAS 부위와 겹치며, 이와 관련된 단백질들은 특정 경로에서 유의미하게 풍부하게 나타나 질병 GWAS 부위의 메커니즘을 규명하는 데 새로운 단서를 제공합니다.
시스 및 트랜스 조절에 따른 질병 표현형의 차이
연구진은 FinnGen 프로젝트에서 얻은 700개 이상의 질병 데이터 세트와 300개의 cis-pQTL 기반 단백질-질병 연관성을 결합했습니다. 그 결과, 이 중 73개만이 멘델 무작위화(MR)와 유전적 위험 신호를 모두 포착하는 통계적 공동 위치 분석 결과를 나타냈습니다.이는 잠재적인 질병 유발 유전자의 우선순위를 정할 때 보완적인 증거가 필요하다는 것을 시사합니다.
평가 가능한 115개의 연관성 중 31개는 일관된 시스 및 트랜스 조절 효과를 보였고, 41개는 이를 뒷받침하는 증거가 없었으며, 14개는 정반대의 효과를 보여 시스 근접 조절과 트랜스 원거리 조절이 질병 표현형에 미치는 영향에 상당한 차이가 있음을 나타냈습니다.
유전적 추론 및 관찰 연구에서의 단백질-질병 연관성 분석
본 연구는 UKBB 연구 참가자 52,164명의 관찰 데이터와 PanBio 데이터베이스에 있는 129만 명 이상의 유전 데이터를 통합하여 517개 질환을 분석했습니다. 신뢰도가 높은 193개의 유전적 연관성 중 관찰 연구에서 일관되게 뒷받침된 것은 52개에 불과했으며, 유의미한 관찰 연관성 52,887개 중 유전적 증거를 확보한 것은 TP3T 유전자 0.061개뿐이었습니다. 특히, 혈액 푸린 단백질은 유전 연구와 관찰 연구 모두에서 고혈압, 심근경색, 심방세동과 일관되게 연관된 몇 안 되는 표적 중 하나로, 신약 개발에 있어 잠재적 가치를 시사합니다.
trans-pQTL은 질병 바이오마커 발굴 및 약물 재표적화에 도움을 줍니다.
901개 이상의 TP3T 질병 단백질 바이오마커(307개 질병 중 280개)가 전사 조절 pQTL 관련 단백질에서 유의미하게 풍부하게 나타났으며, 이는 전사 조절이 질병 단백질 바이오마커의 핵심 유전적 기반임을 확인시켜 줍니다. 본 연구에서는 TYK2 유전자 미스센스 돌연변이 rs34536443이 전사 조절 pQTL로서 BST2 및 CXCL9/10/11과 같은 여러 염증성 단백질을 조절한다는 사실을 발견했습니다. 이러한 단백질 수치 증가는 류마티스 관절염, 건선 및 자가면역 갑상선염의 위험 증가와 관련이 있으며, 이는 자가면역 질환에 대한 TYK2 억제제의 용도 변경에 대한 유전적 근거를 제공합니다.
결론
본 연구는 세계 최대 규모의 다중 코호트 프로테오게놈 분석을 기반으로 인체 순환 프로테옴의 유전적 조절 패턴을 체계적으로 규명했습니다. 기존 연구들이 시스 조절에만 초점을 맞추었던 한계를 뛰어넘어, 대규모 샘플 수준에서 순환 단백질 발현량 조절에 있어 트랜스 유전적 조절의 핵심적인 역할을 최초로 종합적으로 밝혀냈습니다. 나아가 머신러닝 기법을 활용하여 작용 유전자를 정밀하게 찾아내고, N-결합 당화 및 혈소판 생물학 등의 핵심 경로와 간, 면역 세포 등의 주요 조절 부위를 규명했습니다.
이 연구는 단백질체학 기술이 순환 단백질의 일부 아형 및 번역 후 변형만을 포괄한다는 점, 그리고 연구 대상 집단이 유럽계에 국한되어 있어 더 다양한 민족 집단으로 확장될 필요가 있다는 점 등 몇 가지 한계를 지니고 있지만, 비코딩 유전적 변이, 순환 단백질, 질병 메커니즘을 연결하는 완전한 틀을 제시한다는 점에서 의미가 있다. 이는 복잡한 질병의 분자 메커니즘을 규명하는 새로운 관점을 제공할 뿐만 아니라, 혈장 푸린(plasma furin) 및 TYK2와 같은 핵심 표적을 유전적 증거를 통해 규명하여 혁신적인 신약 개발 및 약물 재활용을 위한 매우 신뢰할 수 있는 유전적 근거를 제공하고, 단백질유전체학이 기초 연구에서 임상 적용으로 나아가는 데 중요한 발걸음을 내딛게 한다.








