Command Palette
Search for a command to run...
精製された配列を用いたAlphaFold3による状態認識型タンパク質-リガンド複合体予測
精製された配列を用いたAlphaFold3による状態認識型タンパク質-リガンド複合体予測
Enming Xing Junjie Zhang Shen Wang Xiaolin Cheng
ワンクリックでデプロイ可能なAlphaFold3
概要
タンパク質-リガンド複合体のディープラーニングに基づく予測は、AlphaFold3、Boltz-1、Chai-1、Protenix、NeuralPlexerなどのアーキテクチャの開発に伴い、著しく進展している。複数配列アライメント(MSA)は構造推論に不可欠な共進化情報を提供するため、重要な入力として用いられてきた。しかし、最近のベンチマーク研究により、これらのモデルは訓練データからリガンドのポーズを暗記しており、新規な化学構造や結合ポケットにおける大きな立体構造変化を伴う動的な結合事象に対しては性能が低いという重大な限界があることが明らかになっている。この課題を克服するため、我々は、本研究室が以前に開発したAF-ClaSeqという手法によって生成された精製された配列サブセットを活用した、状態認識型タンパク質-リガンド予測戦略を導入した。AF-ClaSeqは共進化シグナルを分離し、AlphaFold2によって予測された異なる立体構造状態を優先的にエンコードする配列を選択する。MSA由来の立体構造制約を適用した結果、リガンドのポーズ予測において顕著な改善が観察された。AlphaFold3が失敗し、誤ったリガンド配置および関連するタンパク質の立体構造を生成していたケースにおいて、関連する機能状態(例えば、負のアルロステリックモジュレーターと結合した酵素の不活性型)に対応する配列サブセットを使用することで、予測の修正に成功した。
One-sentence Summary
This study introduces a state-aware protein-ligand complex prediction strategy that integrates AlphaFold3 with AF-ClaSeq purified sequence subsets to isolate coevolutionary signals for distinct structural states, applying MSA-derived conformational restraints to overcome ligand pose memorization and correct AlphaFold3 prediction failures in dynamic binding events.
Key Contributions
- A state-aware protein-ligand prediction strategy integrates AF-ClaSeq-derived purified sequence subsets into deep learning structure prediction pipelines. This approach filters multiple sequence alignments to retain only sequences encoding distinct structural conformations, applying evolutionary restraints that guide folding algorithms toward ligand-compatible states.
- Accurate conformational sampling depends on sequence purity rather than alignment depth, with state-specific evolutionary signals distributed across diverse phylogenetic clades. This finding circumvents the signal-averaging limitations of attention-based transformers when processing heterogeneous evolutionary data.
- Incorporating purified sequence subsets into the AlphaFold3 framework produces more accurate ligand placements and binding pocket geometries than default predictions. The method corrects prior modeling failures on targets requiring substantial conformational rearrangements or novel chemotype binding.
Introduction
Accurate prediction of protein-ligand complexes is a cornerstone of computational drug discovery and molecular modeling, with recent deep learning architectures like AlphaFold3 achieving breakthrough accuracy by leveraging coevolutionary signals from multiple sequence alignments. Despite these advances, current models frequently struggle with novel chemotypes and fail to capture binding pocket plasticity, often defaulting to a single static conformation or relying on memorized structural patterns from training data. To overcome these barriers, the authors leverage a sequence purification framework called AF-ClaSeq to isolate evolutionary subsets that specifically encode distinct protein conformations. By integrating these state-specific sequence subsets into AlphaFold3, they introduce conformational restraints that guide the model toward functionally relevant structural states, significantly improving ligand pose accuracy and enabling reliable predictions for dynamic binding events.
Method
The authors leverage a sequence purification strategy to improve the prediction of protein-ligand complexes involving allosteric inhibitors, particularly for proteins like EGFR and IL-1β where the target conformation is distinct from the default state predicted by AlphaFold3 (AF3). The core of the method involves biasing the multiple sequence alignment (MSA) used by AF3 to favor a specific protein conformation, thereby guiding the model toward the desired structural state. The process begins with a deep MSA generated from a large pool of homologous sequences. For EGFR, the initial MSA of 49,743 sequences was filtered to 43,365 using a coverage threshold. To assess the conformational bias encoded within this MSA, the authors performed a series of M-fold sampling experiments, predicting structures from randomly shuffled groups of sequences. The initial analysis revealed a strong bias toward the active state, which was attributed to the model's tendency to predict well-represented conformations.
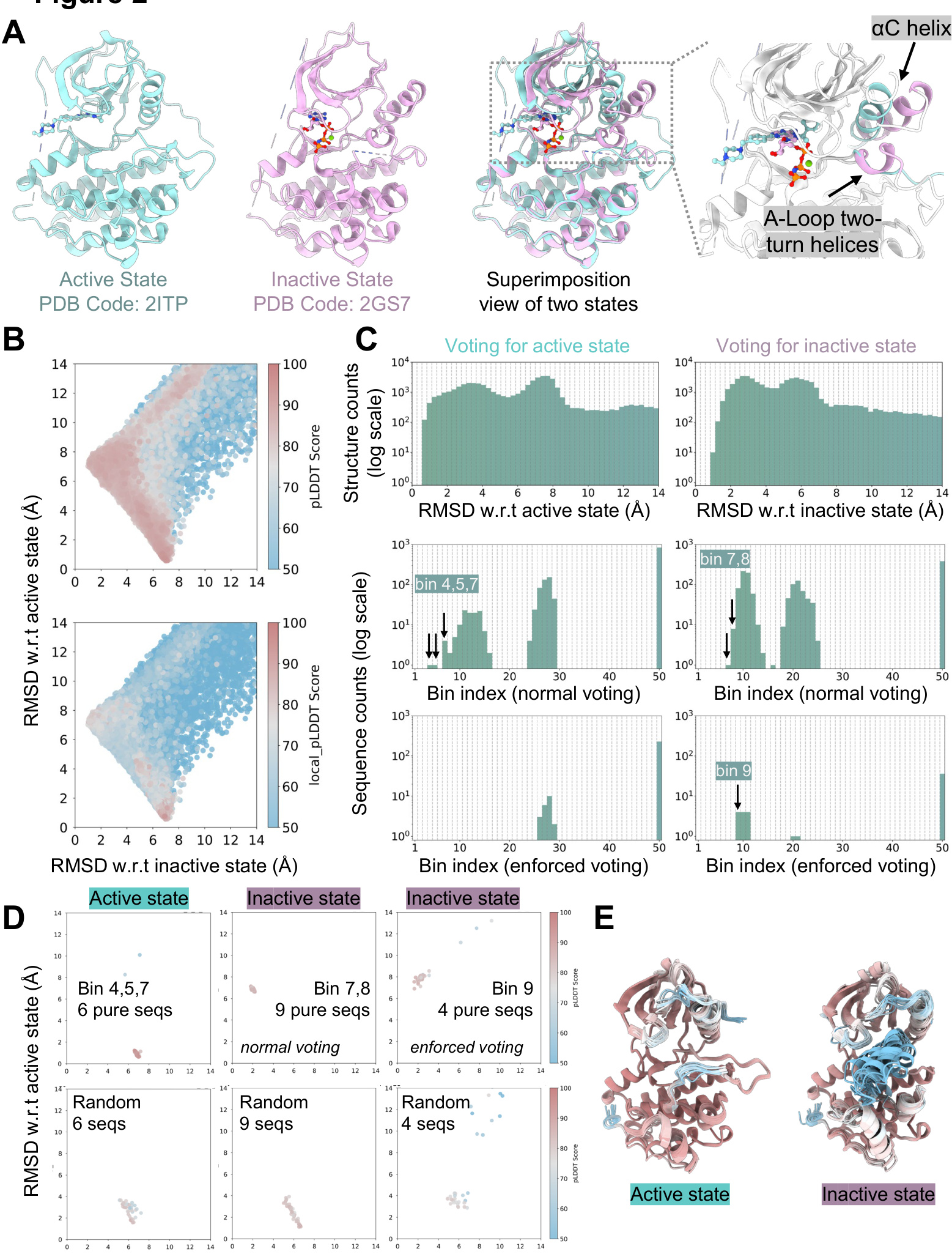
To correct this bias, an iterative enrichment procedure was applied. The method uses the root-mean-square deviation (RMSD) of key structural elements—the αC helix and activation loop (A-Loop)—relative to a reference structure of the desired inactive state (PDB 2GS7) as a metric. Sequences are grouped into sets of six, and after prediction, the sequences yielding structures with the lowest RMSD to the inactive state are selected for the next iteration. This process was repeated four times, progressively enriching the sequence pool for those encoding the inactive conformation. The enriched sequence set from the final iteration was then used for M-fold sampling, generating a large number of predictions to map the conformational landscape. The results, visualized in a scatter plot, show a distribution of predicted structures along a conformational axis, with the local pLDDT score indicating the confidence of the predicted secondary structures.
To identify the most strongly biased sequences, a voting mechanism was employed. The prediction results were binned based on their RMSD to the active and inactive states. A "normal voting" scheme selected sequences that were most frequent in a given bin. A more stringent "enforced voting" scheme required not only the highest frequency but also that the frequency exceeded a threshold of 0.15, isolating sequences with a very strong conformational preference. This process identified a subset of sequences, designated as "pure sequences," that were highly biased toward the inactive state. These purified sequences were used to generate a final MSA for AF3 predictions of the protein-ligand complex, with the ligand's SMILES provided as input.
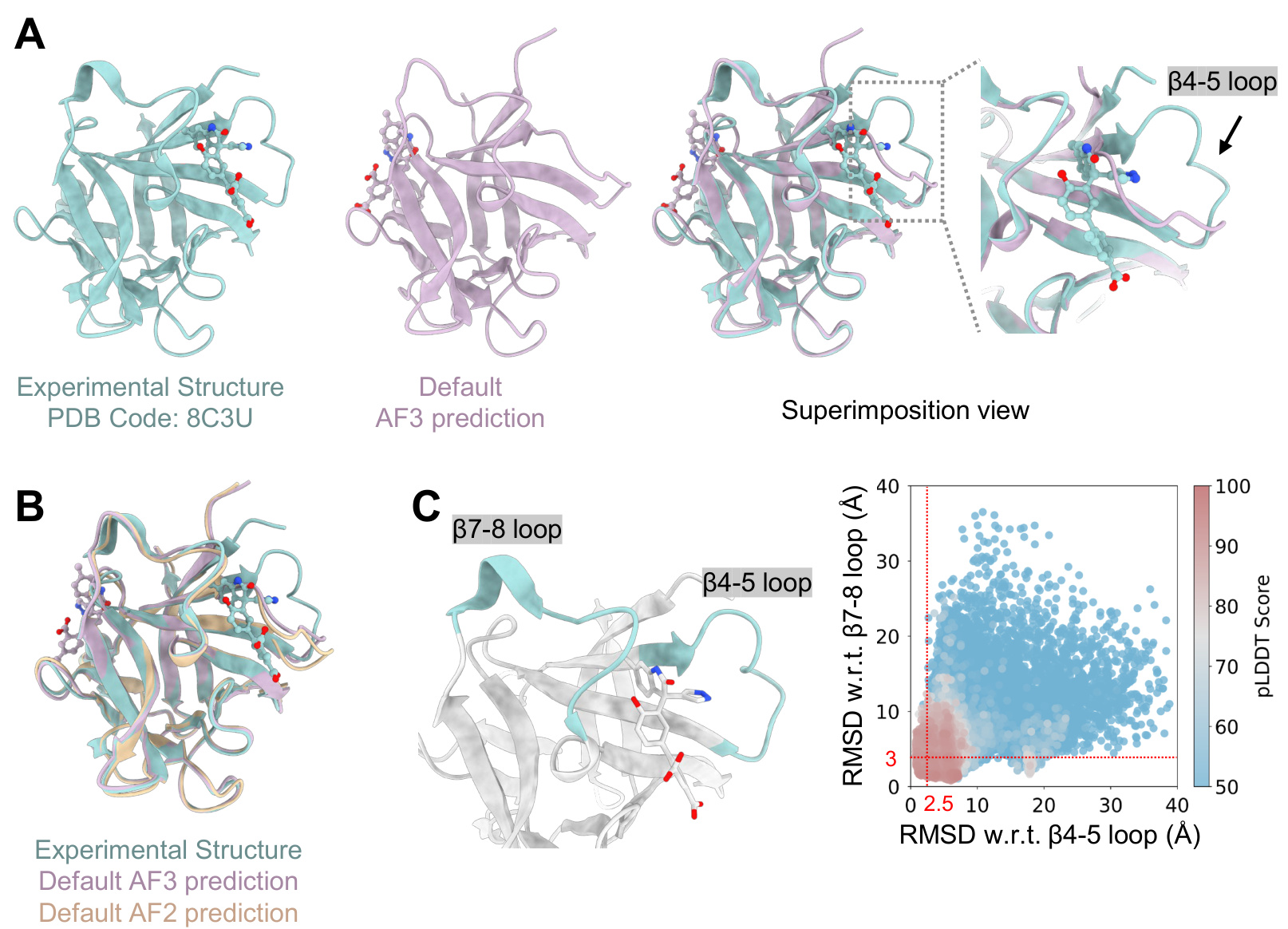
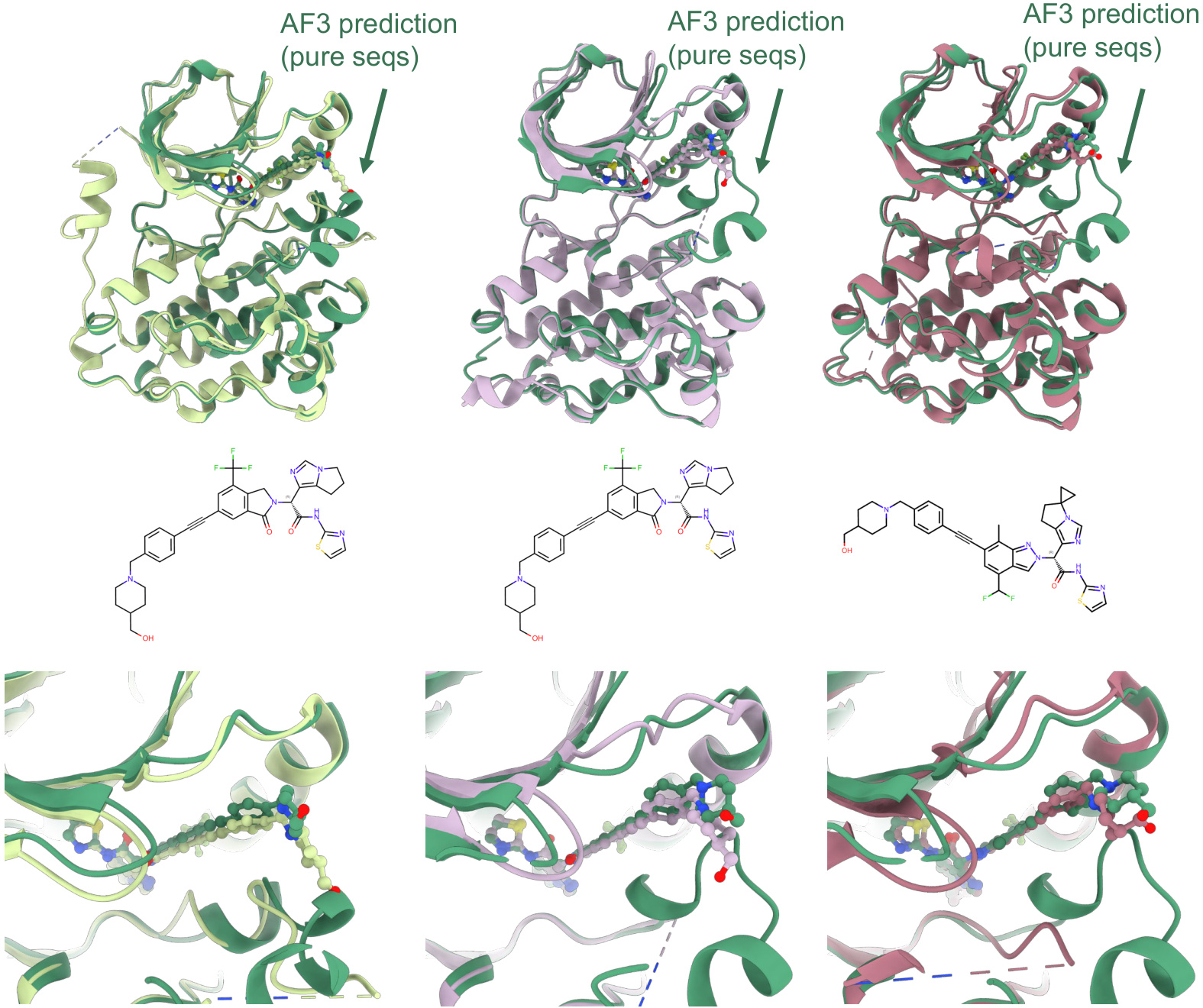
The method was validated on the EGFR-4 allosteric inhibitor complexes. Default AF3 predictions were highly divergent and failed to reproduce the correct ligand pose. However, predictions using the purified inactive-state sequences showed dramatically improved accuracy and consistency. The ligand RMSD dropped to near or below 2.5 Å, and the ligand atomic pLDDT scores increased significantly, indicating a reliable prediction of the binding mode. A similar approach was applied to the IL-1β/ligand system, where the key conformational change involves the displacement of the β4-5 loop. A similar iterative enrichment based on RMSD to the displaced loop state was used to generate an enriched sequence set. Predictions using the top 20 most frequent sequences from the target conformational region achieved perfect alignment with the experimental structure, demonstrating the power of conformational restraints derived from the MSA.
Experiment
The study evaluated two allosteric inhibitor systems, EGFR mutants and IL-1β cryptic pocket antagonists, by comparing default AlphaFold3 predictions against those guided by iterative sequence purification based on generalized conformational references. While default models consistently failed to capture the necessary allosteric or cryptic binding conformations and tended to revert to memorized training patterns, the purification approach successfully biased multiple sequence alignments toward functionally relevant inactive or allosteric states. Consequently, the refined predictions closely matched experimental crystal structures for both protein backbones and ligand poses, demonstrating that conformational constraint-guided sequence enrichment provides a broadly applicable framework for accurately modeling novel allosteric drug-target interactions.