Command Palette
Search for a command to run...
ベストを打ち勝つ:タンパク質構造予測におけるAlphaFold2の改善
ベストを打ち勝つ:タンパク質構造予測におけるAlphaFold2の改善
Abbi Abdel-Rehim Oghenejokpeme Orhobor Hang Lou Hao Ni Ross D. King
ワンクリックで AlphaFold2 をデプロイ
概要
タンパク質構造予測(PSP)問題の目的は、アミノ酸配列からタンパク質の三次元構造を予測することである。アンフィンセンによるノーベル賞受賞研究がタンパク質のコンフォメーションが配列によって決定されることを示して以来、この問題は科学における「聖杯」とされてきた。この目標に向けた最近の重要な進展は、現在最も優れたPSP手法であるAlphaFold2の開発であった。AlphaFold2は、おそらく科学分野におけるAI応用の中で最も注目度の高いものである。AlphaFold2およびRoseTTAFold(もう一つの印象的なPSP手法)の両方は、論文として発表され、コードおよびモデルが公開されている。スタッキングは、アンサンブル機械学習(ML)の一形態であり、複数のベースラインモデルをまず学習し、その後、ベースラインレベルモデルの出力を用いてメタモデルを学習することで、ベースモデルを上回る性能を持つモデルを構築する。スタッキングは多くの応用で成功を収めている。我々は、AlphaFold2とRoseTTAFoldをスタッキングすることにより、ARStackというPSP手法を開発した。ARStackはAlphaFold2を大幅に上回る性能を示す。このことを、非相同なタンパク質の2つのセット、およびAlphaFold2およびRoseTTAFoldの公開後に発表されたタンパク質構造のテストセットを用いて厳密に実証した。より高品質な予測手法が次々と発表されるにつれて、アンサンブル手法が単一手法を凌駕するケースがますます増えていくと考えられる。
One-sentence Summary
The authors introduce ARStack, a protein structure prediction method that employs stacking ensemble learning to integrate AlphaFold2 and RoseTTAFold, significantly outperforming AlphaFold2 on two non-homologous protein sets and a test set of structures published after the base models.
Key Contributions
- This work introduces ARStack, a stacking ensemble method that combines AlphaFold2 and RoseTTAFold as baseline predictors. A neural network meta-model is trained to learn the mapping between baseline predictions and true protein conformations.
- The ensemble architecture significantly outperforms the standalone AlphaFold2 model in predicting three-dimensional protein structures.
- Evaluation employs two sets of non-homologous proteins and a test set of structures published after the baseline models. This benchmarking confirms the generalization capability of the stacking approach on unseen targets.
Introduction
Predicting a protein's three-dimensional structure from its amino acid sequence remains a foundational challenge in computational biology because structural accuracy directly enables functional insights and drug discovery. While deep learning models like AlphaFold2 and RoseTTAFold have dramatically advanced the field, each exhibits complementary strengths and occasional inaccuracies that limit standalone reliability. To address this, the authors leverage stacking ensemble machine learning to combine these two top-tier predictors into ARStack, a neural network meta-model that synthesizes their individual outputs. By training on diverse test sets, including proteins released after the baseline models, ARStack consistently outperforms AlphaFold2 alone, demonstrating that ensemble strategies can reliably push prediction accuracy beyond the current state of the art.
Dataset
-
Dataset Composition and Sources: The authors construct three protein datasets sourced from the RCSB PDB, filtering for structures with at least 50 amino acids and unique Uniprot accession numbers. Proteins are organized into homologous sets using UniProtKB family annotations. Datasets 1 and 2 consist of human proteins selected for their medical relevance, while Dataset 3 contains structures published after the initial release of the structure predictors.
-
Subset Details: Dataset 1 includes one randomly selected protein per homologous set. Dataset 2 contains two randomly selected proteins per set. Dataset 3 acts as a temporal holdout to evaluate generalization on newer structures. Complete protein inventories are provided in the supplementary materials.
-
Training Usage and Processing: The authors use these datasets to train and evaluate the ARStack model. Sequence alignment and targeted truncations are applied to ensure exact matches between predicted and experimental sequences. Predicted structures are then superimposed onto experimental structures using singular value decomposition via BioPython, and the resulting transformed coordinates are extracted for downstream tasks.
-
Target Construction and Atom Handling: The authors calculate target values by computing vectors between AlphaFold2 and RoseTTAFold coordinates for each atom. The model is trained to predict the location on each vector that lies closest to the experimental coordinates, with targets defined as the distance and direction to that optimal point. Because RoseTTAFold outputs only backbone atoms, the pipeline processes just these three atoms per residue, expanding the target columns to three times the size of the input descriptors.
Method
The authors leverage a machine learning framework that begins with the construction of feature descriptors derived from multiple sequence alignments (MSAs). The core of this process involves extracting positional information from the Uniref90 MSA to generate a rich representation for each amino acid in a protein chain. For each position, the fractions of the 20 standard amino acids and gap characters present across the aligned sequences are computed, forming a 21-dimensional vector. This vector captures the evolutionary conservation and variability at that site. To incorporate local context, the vector for each position is concatenated with the vectors of the 10 preceding and 10 succeeding positions, resulting in a 431-unit descriptor for the central position. For positions near the termini of the chain, where flanking residues are unavailable, zero-filled vectors are used to maintain consistent dimensionality. 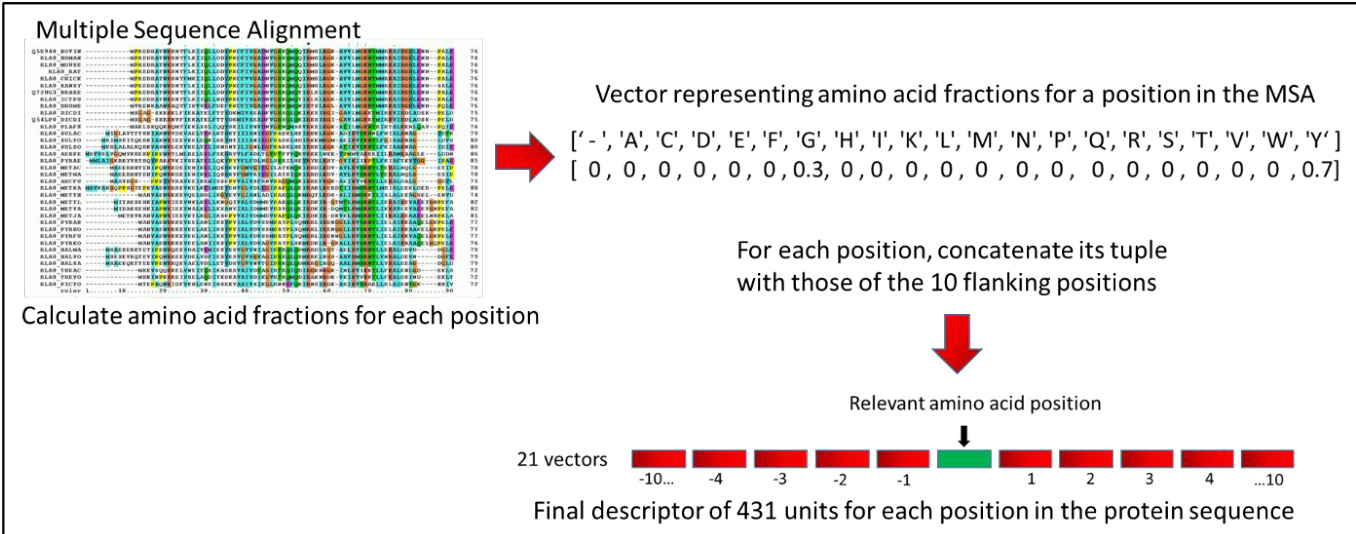
The constructed descriptors serve as input to a deep neural network (DNN) architecture implemented using TensorFlow and Keras. The network consists of four layers: an input layer with 431 neurons, matching the descriptor dimension; three hidden layers containing 200, 200, and 100 neurons respectively; and an output layer with a single neuron to predict the target value. A dropout rate of 0.2 is applied between the hidden layers to mitigate overfitting. The first two hidden layers utilize the rectified linear unit (ReLU) activation function, while the third hidden layer employs softmax, indicating a classification task. The model is trained using stochastic gradient descent with a learning rate of 0.01 and optimized using mean squared error as the loss function. Training is performed over three epochs with a batch size of 150 samples. Model evaluation is conducted using a leave-one-out cross-validation strategy, where each protein is held out in turn for testing. 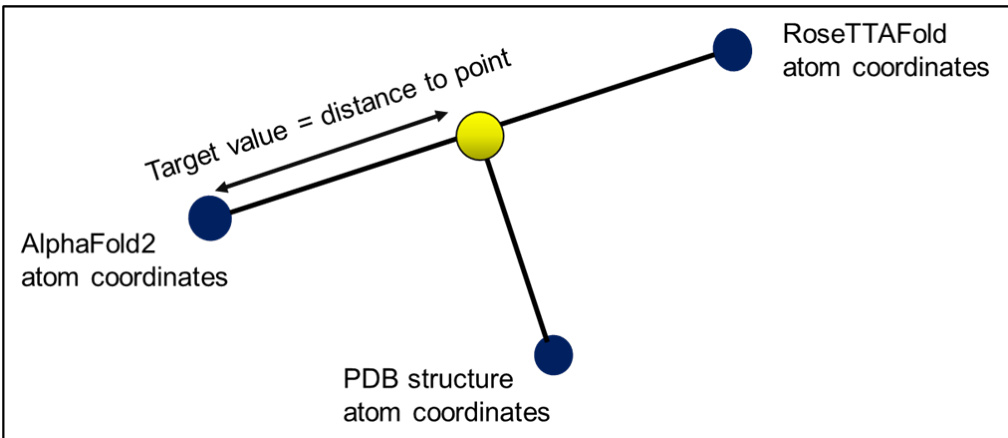
Experiment
The evaluation compares a stacking ensemble method that combines AlphaFold2 and RoseTTAFold against AlphaFold2 alone, utilizing three distinct datasets to validate general prediction accuracy, the impact of homologous training structures, and robustness against training data leakage. Qualitative analysis demonstrates that the stacked approach consistently outperforms the single model, delivering the most reliable improvements for challenging targets and smaller, more flexible proteins. Ultimately, the results indicate that integrating complementary predictors effectively corrects individual model limitations, establishing ensemble-based strategies as a highly promising direction for the future of protein structure prediction.
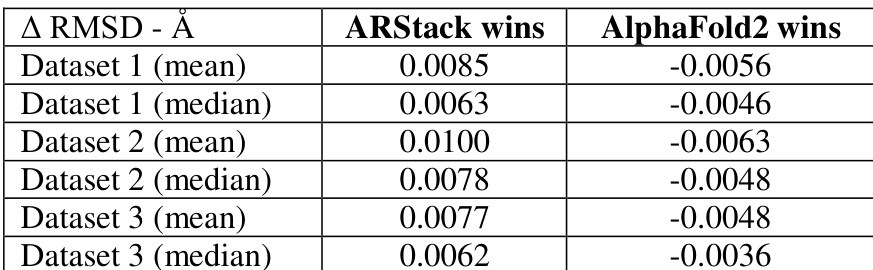
The authors compare the performance of ARStack against AlphaFold2 across three datasets, showing that ARStack consistently achieves better results in terms of RMSD improvements. The analysis reveals that ARStack outperforms AlphaFold2 in most cases, with more significant gains observed in proteins where AlphaFold2 predictions are less accurate, particularly for smaller proteins. The results are statistically significant across all datasets, supporting the effectiveness of the stacking approach. ARStack consistently achieves better RMSD improvements compared to AlphaFold2 across all datasets. ARStack provides more pronounced improvements for proteins where AlphaFold2 predictions are less accurate. The benefits of ARStack are more pronounced for smaller proteins, with higher win rates in lower molecular weight ranges.
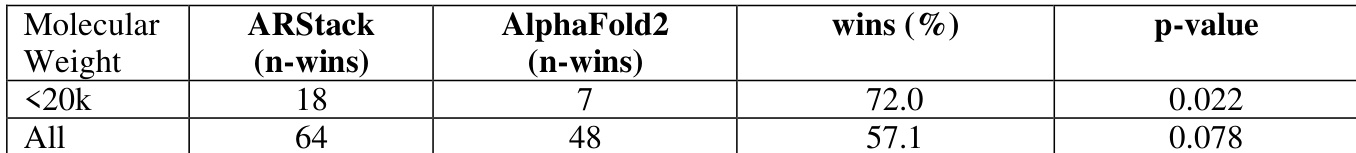
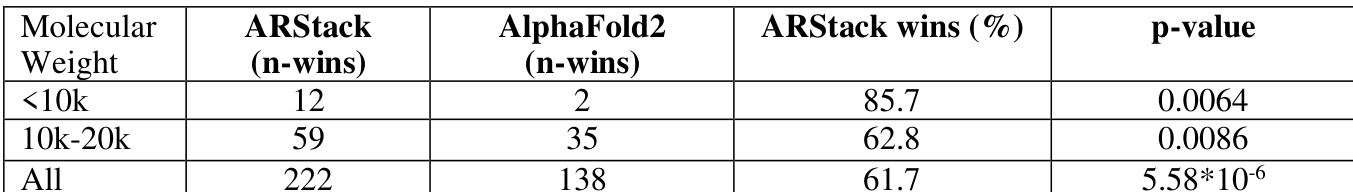
The authors analyze the performance of ARStack compared to AlphaFold2 across different molecular weight ranges, finding that ARStack outperforms AlphaFold2 more consistently in smaller proteins. The results indicate a higher success rate for ARStack in lower molecular weight categories, with statistically significant improvements observed. ARStack shows a higher win rate than AlphaFold2 in proteins with molecular weight below 20k. The performance advantage of ARStack becomes more pronounced in smaller proteins, with a statistically significant improvement observed. ARStack consistently outperforms AlphaFold2 across all molecular weight categories, with the largest relative benefit in the smallest proteins.
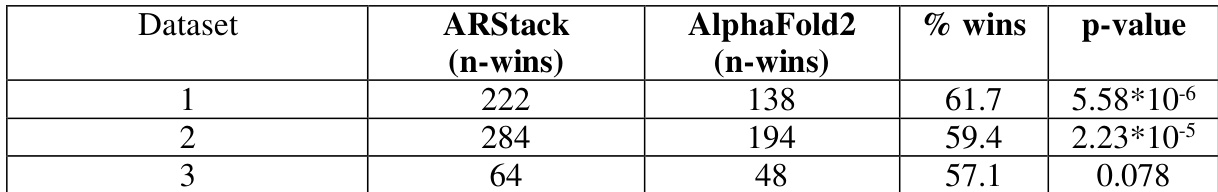
The authors compare the performance of ARStack and AlphaFold2 across multiple datasets, finding that ARStack consistently outperforms AlphaFold2 in terms of structural prediction accuracy. The improvement is particularly notable in smaller proteins and when AlphaFold2 predictions are less accurate, with ARStack providing greater corrections in these cases. ARStack consistently outperforms AlphaFold2 across all datasets, with improvements more pronounced in smaller proteins and when AlphaFold2 predictions are less accurate. The benefits of ARStack are most significant in cases where AlphaFold2 predictions are poor, as indicated by larger initial RMSD values. Protein size and flexibility are correlated with the effectiveness of ARStack, with smaller and more flexible proteins showing greater improvement from the stacking approach.
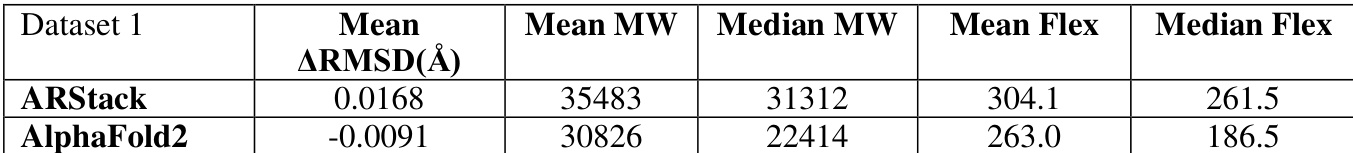
The authors analyze the performance of ARStack compared to AlphaFold2 across different protein molecular weight ranges, showing that ARStack consistently outperforms AlphaFold2, with a stronger advantage observed in smaller proteins. The results indicate that the improvement from stacking is more pronounced in proteins below 10kDa, where ARStack wins in a significantly higher percentage of cases. Statistical analysis confirms the significance of these findings, particularly for smaller proteins. ARStack outperforms AlphaFold2 across all molecular weight ranges, with a more pronounced advantage in smaller proteins. The improvement from ARStack is most significant for proteins below 10kDa, where it wins in a substantially higher percentage of cases. Statistical analysis confirms the significance of ARStack's superior performance, especially in smaller proteins.
The authors compare ARStack, a stacking method combining AlphaFold2 and RoseTTAFold, with AlphaFold2 across three datasets. Results show that ARStack outperforms AlphaFold2 in a majority of cases, with statistically significant improvements on two datasets and a trend toward significance on the third. The performance gain is more pronounced for smaller proteins and when AlphaFold2 predictions are less accurate. ARStack outperforms AlphaFold2 on all three datasets, with statistically significant improvements on two datasets. The performance gain of ARStack is more pronounced for smaller proteins and when AlphaFold2 predictions are less accurate. ARStack shows a higher percentage of wins compared to AlphaFold2, with the difference being most significant on Dataset 1.
The experiments evaluate the stacking method ARStack against AlphaFold2 across three datasets, validating the approach through comparative analyses across different molecular weight ranges and baseline accuracy levels. The results demonstrate that ARStack consistently improves structural predictions, with the most substantial gains occurring in smaller, more flexible proteins and cases where the original model performs poorly. These findings confirm that the stacking strategy effectively corrects structural errors and provides robust, statistically significant enhancements across diverse protein targets.