Command Palette
Search for a command to run...
Calcul Fonctionnel Hybride VASP De La Densité d'états Et Des Bandes d'énergie Du Silicium
Package de simulation ab initio de Vienne (VASP) Il s'agit d'un programme informatique pour la modélisation de matériaux à l'échelle atomique à partir de principes de base, tels que les calculs de structure électronique et la dynamique moléculaire de la mécanique quantique.
VASP est adapté au calcul de solutions approximatives à l'équation de Schrödinger à plusieurs corps. Il couvre une variété de méthodes de calcul, notamment la résolution de l'équation de Kohn-Sham dans le cadre de la théorie de la fonctionnelle de la densité (DFT) et le traitement de l'équation de Roothaan dans l'approximation Hartree-Fock (HF). Une autre innovation de VASP est qu’il intègre la méthode Hartree-Fock à la théorie de la fonctionnelle de la densité pour former une méthode fonctionnelle hybride, qui offre aux chercheurs des options de calcul plus flexibles. De plus, les méthodes de fonction de Green (quasiparticules GW et ACFDT-RPA) et la théorie des perturbations à plusieurs corps (Møller-Plesset du second ordre) sont disponibles dans VASP.
Dans VASP, les quantités centrales telles que les orbitales électroniques individuelles, la densité de charge électronique et le potentiel local sont représentées à l'aide d'ensembles de base d'ondes planes. Les interactions entre électrons et ions sont décrites à l'aide de méthodes de conservation de la norme ou de pseudopotentiels ultra-doux ou d'ondes augmentées par projecteur.
Pour déterminer l'état fondamental électronique, VASP utilise des techniques efficaces de diagonalisation de matrice itérative telles que la méthode de minimisation résiduelle avec sous-espace itéré à inversion directe (RMM-DIIS) ou l'algorithme de Davidson par blocs. Ceux-ci sont combinés avec des schémas de mélange de densité Broyden et Pulay efficaces pour accélérer la boucle auto-cohérente.
Introduction au tutoriel
Ce tutoriel vous apprendra à calculer les bandes d'énergie du silicium à l'aide de fonctionnelles hybrides, ce qui est similaire au tutoriel précédent (Tutoriel de démarrage VASP : Calcul de la densité d'états et des bandes passantes dans le silicium), la fonctionnelle hybride peut calculer les bandes d'énergie et la densité d'état du matériau avec la bande interdite correcte, mais elle nécessite plus de ressources de calcul, donc le GPU est utilisé pour la démonstration cette fois.
En suivant ce tutoriel, vous apprendrez à générer des INCAR et des KPOITNS qui sont nécessaires aux calculs fonctionnels hybrides VASP.
Tutoriel précédent (Tutoriel de démarrage VASP : Calcul de la densité d'états et des bandes passantes dans le silicium) contient 4 étapes. Le calcul de cette fonctionnelle hybride ne nécessite qu'une seule étape (l'optimisation structurelle sera ignorée) pour obtenir la densité d'état et la bande d'énergie du silicium.
Préparation des fichiers d'entrée
Ce tutoriel nécessite la préparation de quatre fichiers : INCAR, POSCAR, KPOINTS et POTCAR.
INCAR
Global Parameters
ENCUT = 300 (波函数截断能量)
PREC = Normal (精度设置)
LWAVE = .TRUE. (保存波函数)
LCHARG = .TRUE. (保存电荷)
ADDGRID= .TRUE. (增加格点加速收敛)
Static Calculation
ISMEAR = 0 (高斯占据数)
SIGMA = 0.1 (高斯展宽)
LORBIT = 11 (输出 DOSCAR 和 PROCAR)
NELM = 60 (最大电子步)
EDIFF = 1E-08 (电子步收敛判据)
HSE06 Calculation
LHFCALC= T (启动杂化泛函计算)
AEXX = 0.25 (杂化比例 0.25)
HFSCREEN= 0.2 (杂化屏蔽参数 0.2)
ALGO = ALL (最优化算法)
TIME = 0.4 (最优化算法步长)
PRECFOCK= N (FFT 精度)
POSCAR
Si #(体系名称)
1.0 #(放大系数 下面 3 行对应 3 个晶格矢量 )
0.0 2.75 2.75
2.75 0.0 2.75
2.75 2.75 0.0
Si #(元素)
2 #(对应元素原子数)
Direct #(采用分数坐标,下列为 2 个原子的分数坐标)
0 0 0
0.25 0.25 0.25
KPOINTS
0.060 5 5 5 10 0.060 83 6 19 6 20 16 13 9 # Parameters to Generate KPOINTS (Do NOT Edit This Line)
93
Reciprocal lattice
0.00000000000000 0.00000000000000 0.00000000000000 1
0.20000000000000 0.00000000000000 0.00000000000000 8
0.40000000000000 0.00000000000000 0.00000000000000 8
0.20000000000000 0.20000000000000 0.00000000000000 6
0.40000000000000 0.20000000000000 0.00000000000000 24
-0.40000000000000 0.20000000000000 0.00000000000000 24
-0.20000000000000 0.20000000000000 0.00000000000000 12
0.40000000000000 0.40000000000000 0.00000000000000 6
-0.40000000000000 0.40000000000000 0.00000000000000 12
-0.40000000000000 0.40000000000000 0.20000000000000 24 (以上为 scf 点位)
0.00000000000000 0.00000000000000 0.00000000000000 0 (以下为能带点位)
0.02777777777778 0.00000000000000 0.02777777777778 0
0.05555555555556 0.00000000000000 0.05555555555556 0
0.08333333333333 0.00000000000000 0.08333333333333 0
0.11111111111111 0.00000000000000 0.11111111111111 0
0.13888888888889 0.00000000000000 0.13888888888889 0
0.16666666666667 0.00000000000000 0.16666666666667 0
0.19444444444444 0.00000000000000 0.19444444444444 0
0.22222222222222 0.00000000000000 0.22222222222222 0
0.25000000000000 0.00000000000000 0.25000000000000 0
0.27777777777778 0.00000000000000 0.27777777777778 0
0.30555555555556 0.00000000000000 0.30555555555556 0
0.33333333333333 0.00000000000000 0.33333333333333 0
0.36111111111111 0.00000000000000 0.36111111111111 0
0.38888888888889 0.00000000000000 0.38888888888889 0
0.41666666666667 0.00000000000000 0.41666666666667 0
0.44444444444444 0.00000000000000 0.44444444444444 0
0.47222222222222 0.00000000000000 0.47222222222222 0
0.50000000000000 0.00000000000000 0.50000000000000 0
0.50000000000000 0.00000000000000 0.50000000000000 0
0.52500000000000 0.05000000000000 0.52500000000000 0
0.55000000000000 0.10000000000000 0.55000000000000 0
0.57500000000000 0.15000000000000 0.57500000000000 0
0.60000000000000 0.20000000000000 0.60000000000000 0
0.62500000000000 0.25000000000000 0.62500000000000 0
0.37500000000000 0.37500000000000 0.75000000000000 0
0.35526315789474 0.35526315789474 0.71052631578947 0
0.33552631578947 0.33552631578947 0.67105263157895 0
0.31578947368421 0.31578947368421 0.63157894736842 0
0.29605263157895 0.29605263157895 0.59210526315789 0
0.27631578947368 0.27631578947368 0.55263157894737 0
0.25657894736842 0.25657894736842 0.51315789473684 0
0.23684210526316 0.23684210526316 0.47368421052632 0
0.21710526315789 0.21710526315789 0.43421052631579 0
0.19736842105263 0.19736842105263 0.39473684210526 0
0.17763157894737 0.17763157894737 0.35526315789474 0
0.15789473684211 0.15789473684211 0.31578947368421 0
0.13815789473684 0.13815789473684 0.27631578947368 0
0.11842105263158 0.11842105263158 0.23684210526316 0
0.09868421052632 0.09868421052632 0.19736842105263 0
0.07894736842105 0.07894736842105 0.15789473684211 0
0.05921052631579 0.05921052631579 0.11842105263158 0
0.03947368421053 0.03947368421053 0.07894736842105 0
0.01973684210526 0.01973684210526 0.03947368421053 0
0.00000000000000 0.00000000000000 0.00000000000000 0
0.00000000000000 0.00000000000000 0.00000000000000 0
0.03333333333333 0.03333333333333 0.03333333333333 0
0.06666666666667 0.06666666666667 0.06666666666667 0
0.10000000000000 0.10000000000000 0.10000000000000 0
0.13333333333333 0.13333333333333 0.13333333333333 0
0.16666666666667 0.16666666666667 0.16666666666667 0
0.20000000000000 0.20000000000000 0.20000000000000 0
0.23333333333333 0.23333333333333 0.23333333333333 0
0.26666666666667 0.26666666666667 0.26666666666667 0
0.30000000000000 0.30000000000000 0.30000000000000 0
0.33333333333333 0.33333333333333 0.33333333333333 0
0.36666666666667 0.36666666666667 0.36666666666667 0
0.40000000000000 0.40000000000000 0.40000000000000 0
0.43333333333333 0.43333333333333 0.43333333333333 0
0.46666666666667 0.46666666666667 0.46666666666667 0
0.50000000000000 0.50000000000000 0.50000000000000 0
0.50000000000000 0.50000000000000 0.50000000000000 0
0.50000000000000 0.47916666666667 0.52083333333333 0
0.50000000000000 0.45833333333333 0.54166666666667 0
0.50000000000000 0.43750000000000 0.56250000000000 0
0.50000000000000 0.41666666666667 0.58333333333333 0
0.50000000000000 0.39583333333333 0.60416666666667 0
0.50000000000000 0.37500000000000 0.62500000000000 0
0.50000000000000 0.35416666666667 0.64583333333333 0
0.50000000000000 0.33333333333333 0.66666666666667 0
0.50000000000000 0.31250000000000 0.68750000000000 0
0.50000000000000 0.29166666666667 0.70833333333333 0
0.50000000000000 0.27083333333333 0.72916666666667 0
0.50000000000000 0.25000000000000 0.75000000000000 0
0.50000000000000 0.25000000000000 0.75000000000000 0
0.50000000000000 0.21875000000000 0.71875000000000 0
0.50000000000000 0.18750000000000 0.68750000000000 0
0.50000000000000 0.15625000000000 0.65625000000000 0
0.50000000000000 0.12500000000000 0.62500000000000 0
0.50000000000000 0.09375000000000 0.59375000000000 0
0.50000000000000 0.06250000000000 0.56250000000000 0
0.50000000000000 0.03125000000000 0.53125000000000 0
0.50000000000000 0.00000000000000 0.50000000000000 0
POTCAR
Le système correspond à la combinaison pseudopotentielle d'éléments, ici il s'agit du pseudopotentiel de Si.
Étapes de course
Passons maintenant à la partie pratique du tutoriel. Le tutoriel a préparé les packages requis, vous pouvez donc cloner directement le conteneur. Ce didacticiel utilise les ressources de calcul RTX 4090, l'image de version vasp 6.3.0-cuda11.8 et fonctionne dans l'espace de travail.
1. Cloner et démarrer le conteneur
1.1 Attendez que le conteneur soit chargé, puis cliquez sur Ouvrir l'espace de travail
1.2 Ouvrir le terminal
1.3 Entrez le répertoire wfl
cd tutorials/wfl
1.4 Téléchargez le pseudopotentiel de silicium préparé dans le répertoire wfl
Ici vous pouvez utiliserExemple de fichier du site Web officielPOTCAR pseudopotentiel
2. Exécutez VASP
mpirun -n 1 vasp_std
3. Installez vaspkit
Retourner au répertoire précédent
cd ..
3.1 Installer les dépendances Python
pip install numpy scipy matplotlib
3.2 Configurer vaspkit
chmod 777 setupvk.sh
./setupvk.sh
source ~/.bashrc
cd tutorials
4. Traitement des données à l'aide de vaspkit
4.1 Tracé de la densité d'états et des diagrammes de bandes
Entrez dans le répertoire wfl en tapant :
cd wfl
Entrez la commande pour générer des données de densité d’état :
vaspkit
111
1
Entrez la commande pour utiliser vaspkit pour traiter les données de bande et les tracer :
vaspkit
252
2
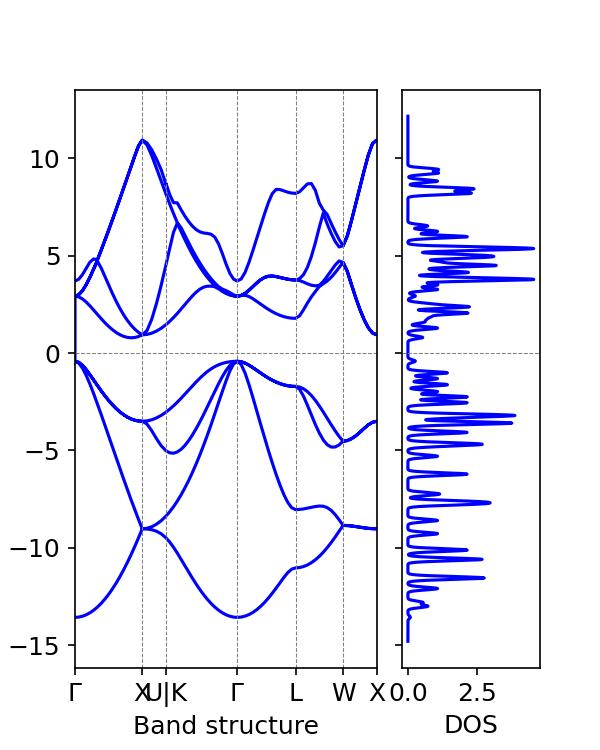
La densité d'états et le diagramme de bande peuvent être générés :
Utilisons vaspkit pour visualiser la bande interdite :
vaspkit
911
On peut obtenir que la bande interdite estimée du silicium est de 1,2 eV, ce qui est proche de la valeur expérimentale typique de la bande interdite du silicium de 1,12 eV.
Dans le tutoriel précédent, les calculs DFT utilisant vaspkit ont révélé une bande interdite de 0,6133 eV, ce qui est loin de la valeur expérimentale.
Par conséquent, les fonctionnelles hybrides peuvent calculer la bande interdite des matériaux avec plus de précision, mais elles nécessitent davantage de ressources de calcul.
Créer de l'IA avec l'IA
De l'idée au lancement — accélérez votre développement IA avec le co-codage IA gratuit, un environnement prêt à l'emploi et le meilleur prix pour les GPU.