Command Palette
Search for a command to run...
One-click Deployment of HiDiffusion
Abstract
One-sentence Summary
FastRemap is a high-performance tool that rapidly remaps genomic sequencing reads between reference assemblies, achieving up to a 7.82× speedup and reducing peak memory consumption to as low as 61.7% compared to the established CrossMap utility.
Key Contributions
- FastRemap is introduced as a high-performance C++ implementation designed to remap genomic read alignments between similar reference genomes.
- The tool modifies the core BAM/SAM remapping logic to address performance bottlenecks and ensure immediate output compatibility with standard downstream analysis pipelines.
- Evaluations across human, C. elegans, and yeast reference genomes demonstrate an average 6.47× speedup and an average 80.7% peak memory consumption relative to CrossMap.
Introduction
As genomic datasets expand and reference assemblies are frequently updated, researchers must efficiently translate existing read mappings to new references. Remapping leverages conserved genomic regions to bypass the computational cost of full re-alignment, making it essential for maintaining high-throughput genome analysis pipelines. Despite its widespread adoption, CrossMap, the current standard remapping tool, struggles with significant performance bottlenecks and produces output formats that break compatibility with downstream variant callers and analysis software. The authors introduce FastRemap, a highly optimized C++ implementation that accelerates the remapping process while ensuring immediate compatibility with standard downstream tools. By refining the underlying algorithms and data handling, the tool delivers up to a 7.82x speedup and reduces peak memory consumption to roughly 62 percent of CrossMap, enabling faster and more reliable genome analysis at scale.
Method
The authors leverage a modular and extensible framework for remapping genomic reads from a source reference to a target reference, building upon the design principles of CrossMap while introducing significant performance and functional improvements. At the core of the system is the use of a chain file, which defines regions of sequence similarity between the source and target references. These chain files are obtained from the UCSC genome browser and are specific to each reference pair, enabling accurate coordinate transformation. The architecture is designed to support multiple data formats, with current implementation focused on SAM/BAM and BED files, while future extensions aim to include VCF, GTF/GFF, BigWig, and MAF formats. This extensibility is facilitated by a clean codebase structure inspired by CrossMap.
For efficient genomic data manipulation, the system relies on the Seqan2 library, which provides robust support for sequence and alignment operations. Additionally, the zlib library is integrated to handle compressed file formats, particularly for BAM input and output, ensuring compatibility with standard genomic data workflows. The implementation is written in C++, which enables high-performance computation, a key advantage over existing tools.
As shown in the figure below:
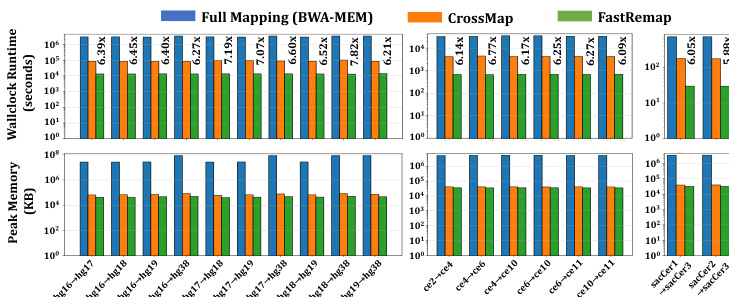
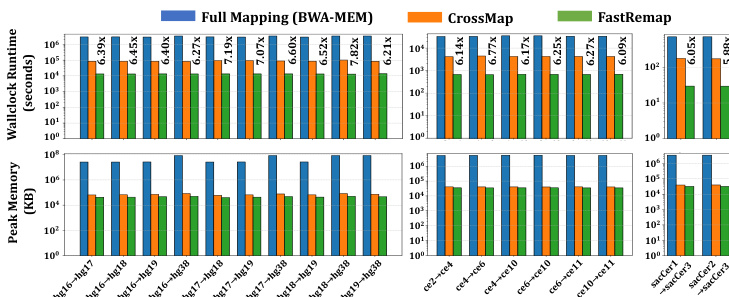
the authors evaluate the performance of FastRemap against BWA-MEM and CrossMap in terms of wall clock runtime and peak memory usage. The results demonstrate that FastRemap achieves substantial speedups over CrossMap, particularly in large-scale datasets, while maintaining low memory footprint. This performance gain is achieved through optimized data structures and efficient chain file traversal algorithms.
Two critical improvements over the original CrossMap implementation are introduced to ensure compatibility with downstream analysis pipelines. First, FastRemap correctly preserves the supplementary alignment flag during remapping of BAM/SAM files. In contrast to CrossMap, which removes this flag, FastRemap maintains accurate alignment metadata, allowing downstream tools such as Picard's MarkDuplicates to process the output without failure. This ensures that the remapped data remains fully compatible with standard variant calling and quality control workflows.
Second, FastRemap generates a separate BED file containing reads that fail to remap. This output enables users to quickly identify and analyze unmappable reads without storing them in the main BAM file, thereby reducing disk space usage and simplifying post-processing. This design choice enhances usability and supports more efficient data management in large-scale sequencing projects.
Experiment
The evaluation compares FastRemap against the leading CrossMap tool and full de novo mapping with BWA-MEM across multiple reference genomes and public DNA sequencing datasets on a high-performance server. By measuring end-to-end execution time and peak memory consumption, the experiments validate the tool's computational efficiency and scalability for genomic read remapping. The qualitative results demonstrate that FastRemap delivers substantially faster processing speeds and markedly reduced memory requirements compared to both competing approaches. Ultimately, the study establishes FastRemap as a highly efficient and scalable solution for modern genomic analysis pipelines.
The authors evaluate FastRemap against CrossMap and full mapping with BWA-MEM, comparing wallclock runtime and peak memory usage across different reference genome pairs for human, C. elegans, and yeast. Results show that FastRemap achieves faster execution and lower memory consumption than both CrossMap and BWA-MEM, with notable improvements in speed and memory efficiency. FastRemap outperforms CrossMap in both runtime and memory usage, while also significantly reducing resource demands compared to full mapping with BWA-MEM. FastRemap achieves faster wallclock runtime and lower peak memory usage compared to CrossMap. FastRemap is significantly faster and more memory efficient than full mapping with BWA-MEM. FastRemap outperforms CrossMap in both speed and memory footprint across multiple reference genome pairs.
The evaluation compares FastRemap against CrossMap and full mapping with BWA-MEM across human, C. elegans, and yeast reference genomes to assess computational efficiency. These experiments validate the practical resource demands of each tool by measuring execution speed and memory consumption. Overall, the findings confirm that FastRemap consistently delivers superior runtime performance and reduced memory overhead, establishing it as a more efficient solution for cross-genome mapping tasks.