Command Palette
Search for a command to run...
dnaHNet:一种用于基因组序列学习的可扩展分层 Foundation Model
dnaHNet:一种用于基因组序列学习的可扩展分层 Foundation Model
Arnav Shah Junzhe Li Parsa Idehpour Adibvafa Fallahpour Brandon Wang Sukjun Hwang Bo Wang Patrick D. Hsu Hani Goodarzi Albert Gu
摘要
基因组基础模型(Genomic foundation models)具有解码 DNA 语法(DNA syntax)的潜力,但在其输入表示(input representation)方面面临着根本性的权衡。传统的固定词表分词器(fixed-vocabulary tokenizers)会将具有生物学意义的模体(motifs)——如密码子(codons)和调控元件(regulatory elements)——进行破碎化处理;而核苷酸级(nucleotide-level)模型虽然能保持生物学连贯性,但在处理长上下文时会产生极高的计算成本。我们推出了 dnaHNet,这是一种最先进的、无需分词器(tokenizer-free)的自回归模型,能够实现基因组序列的端到端分割与建模。通过使用一种可微分的动态分块机制(differentiable dynamic chunking mechanism),dnaHNet 能够自适应地将原始核苷酸压缩为潜在 token,从而在压缩率与预测准确性之间取得平衡。在原核生物基因组上进行预训练后,dnaHNet 在扩展性(scaling)和效率方面均优于包括 StripedHyena2 在内的领先架构。这种递归分块(recursive chunking)带来了二次方级别的 FLOPs 削减,使其推理速度比 Transformer 快 3 倍以上。在零样本(zero-shot)任务中,dnaHNet 在预测蛋白质变异适应性(protein variant fitness)和基因必需性(gene essentiality)方面表现出卓越的性能,同时能够在无需监督的情况下自动发现层级化的生物结构。这些研究结果确立了 dnaHNet 作为下一代基因组建模的可扩展、可解释框架的地位。
一句话总结
通过采用可微动态分块机制将原始核苷酸自适应地压缩为潜在 tokens,无需 tokenizer 的自回归基础模型 dnaHNet 在扩展性和效率方面均优于 StripedHyena2 和 Transformers 等架构,在预测蛋白质变体适应性和基因必需性等 zero-shot 任务上实现了超过 3 倍的推理加速和最先进的性能。
核心贡献
- 本文介绍了 dnaHNet,这是一种无需 tokenizer 的自回归模型,利用可微动态分块机制将原始核苷酸自适应地压缩为潜在 tokens。
- 该架构通过采用递归分块来使 FLOPs 实现平方级降低,从而实现了显著的计算效率,推理速度比 Transformer 模型快 3 倍以上。
- 在原核生物基因组上的实验表明,该模型在扩展性和效率方面优于 StripedHyena2 等领先架构,同时在预测蛋白质变体适应性和基因必需性的 zero-shot 任务中表现出卓越的性能。
引言
基因组基础模型对于解码 DNA 语法以推动药物研发和合成生物学等领域至关重要。然而,当前的方法在计算效率和生物学准确性之间面临着根本性的权衡。固定词表的 tokenizer 虽然高效,但往往会切碎密码子等关键的生物学基序;而核苷酸级模型虽然保留了生物学连贯性,但在处理长基因组上下文时却面临极高的计算成本。通过使用名为 dnaHNet 的分层、无需 tokenizer 的架构,可以解决这一矛盾。通过使用可微动态分块机制,模型将原始核苷酸自适应地压缩为潜在 tokens,使其能够实现卓越的扩展性、更快的推理速度,并在蛋白质变体适应性预测等 zero-shot 任务上达到最先进的性能。
数据集
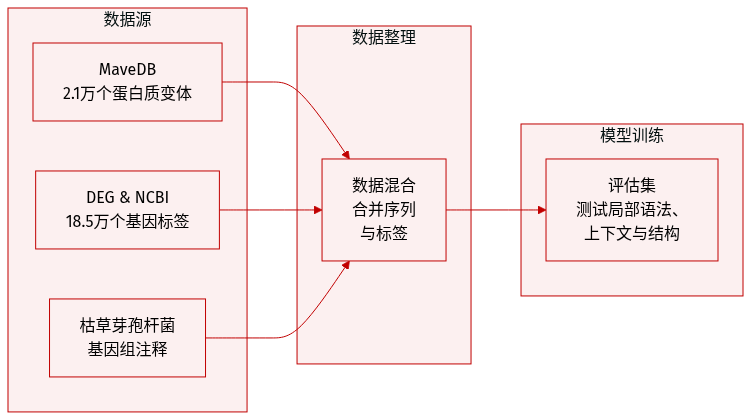
通过使用三个不同的数据集来评估 dnaHNet,这些数据集旨在测试不同的生物学建模能力:
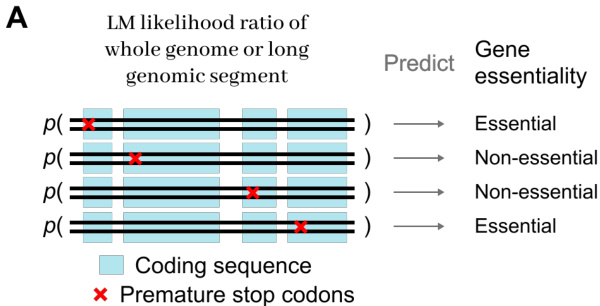
- 蛋白质变体效应 (MaveDB): 该子集由从大肠杆菌 E. coli K-12 的 12 个实验适应性数据集中汇编而成的 21,250 个核苷酸级数据点组成。它用于评估模型捕捉局部编码语法和预测蛋白质适应性景观的能力。
- 基因必需性 (DEG): 该数据集包含 185,226 个数据点,通过从必需基因数据库 (Database of Essential Genes) 中为 62 种细菌生物体的基因生成二进制必需性标签而构建。基础序列和注释来源于 NCBI,如果基因通过名称或序列一致性大于 99% 与 DEG 条目匹配,则将其标记为必需。该子集评估模型整合基因组上下文和长程依赖的能力。
- 基因组结构解释 (NCBI): 为了进行可解释性分析,使用了枯草芽孢杆菌 (B. subtilis) 基因组和来自 NCBI 的功能注释。根据这些注释将基因组划分为不同的功能区域,以确定模型的分割如何与已知的生物学结构对齐。
方法
介绍了 dnaHNet,这是一种专为基因组序列学习设计的、可扩展且无需 tokenizer 的基础模型。该模型将基因组学习建模为一个自回归序列建模问题。给定核苷酸序列 X=(x1,…,xL),其中 xt∈{A,C,G,T},目标是建模概率分布 P(X)=∏t=1LP(xt∣x<t)。为了高效处理长基因组上下文,dnaHNet 利用了由三个主要可微模块组成的递归分层架构:编码器 (E)、主网络 (M) 和解码器 (D)。
参考框架图:
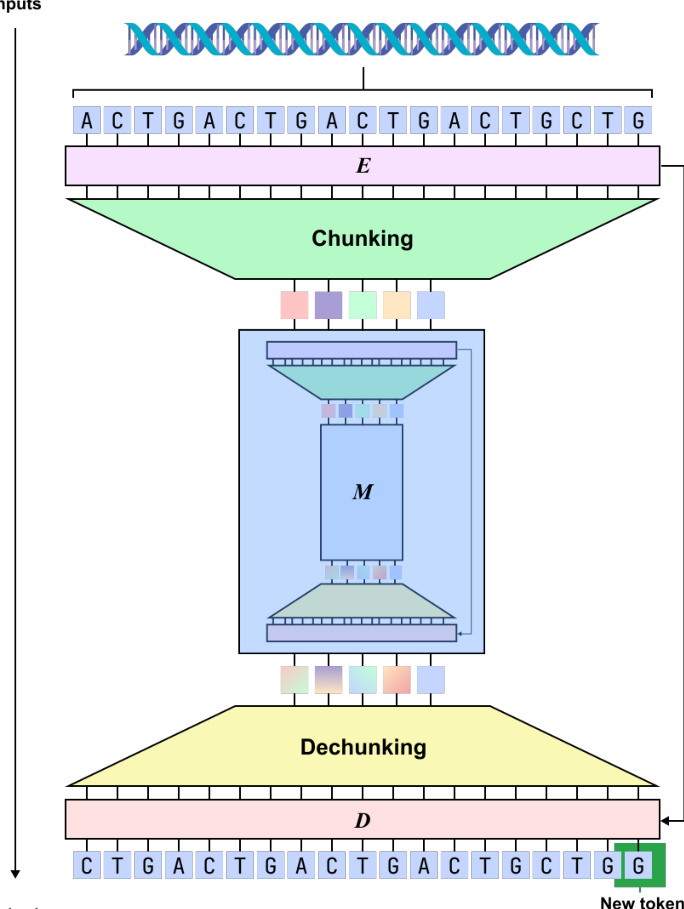
编码器负责通过动态分割机制将核苷酸级输入压缩为潜在分块。它采用了一个由四个 Mamba 层和一个 Transformer 层组成的混合骨干网络。编码器将输入嵌入转换为隐藏状态 h1:L∈RL×D。为了确定分割边界,边界预测模块使用以下公式计算概率 pt∈[0,1]: \npt=21(1−CosineSim(Wqht,Wkht−1)) 其中 Wq 和 Wk 是可学习的投影矩阵。对于表示不相似的核苷酸,会分配较高的边界概率,这鼓励模型在具有生物学意义的转换处(如密码子边界)进行分割。随后,分块层通过选择这些预测边界处的表示来对输出进行下采样,从而得到压缩序列 E=(e1,…,eL′),其中 L′≤L。
如下图所示:
压缩序列 E 随后由主网络 M 处理。该模块可以是标准的 Transformer 或另一个 H-Net 模块,允许通过递归分块来捕获多个抽象层级。在两阶段分层结构中,第一阶段捕获高频局部模式(如密码子周期性),而第二阶段对跨功能区域的长程依赖进行建模。主网络输出处理后的潜在状态 E^=(e^1,…,e^L′)∈RL′×D。
解码器 D 通过两个步骤将这些潜在状态映射回原始核苷酸分辨率。首先,平滑模块使用一种插值离散分块的递归方式,将潜在状态细化为平滑表示 Eˉ=(eˉ1,…,eˉL′): eˉj=Pje^j+(1−Pj)eˉj−1 其中 Pj 是第 j 个分块的边界概率。其次,上采样器通过将向量 eˉc(t) 复制到与分块索引 c(t) 对应的每个核苷酸位置 t,将这些平滑的潜在状态扩展到原始长度 L。随后,解码器利用四个 Mamba 层和一个 Transformer 层来建模自回归依赖,并使用线性头将输出投影到核苷酸词表 logits,以产生下一个核苷酸的分布。
训练过程使用复合目标进行端到端执行。主要部分是自回归 next-token 预测损失: LNLL=−∑t=1LlogPθ(xt∣x<t) 为了防止训练期间出现退化的分割,引入了比例损失,将动态分块正则化为每个阶段 s 的目标下采样率 Rs: Lrate(s)=Rs−1Rs((Rs−1)FsGs+(1−Fs)(1−Gs)) 其中 Fs 是所选分块的实际比例,Gs 是平均边界概率。总损失定义为 L=LNLL+α∑sLrate(s),其中 α 是正则化系数。
实验
实验通过扩展定律分析、zero-shot 生物学预测任务和结构可解释性研究,将 dnaHNet 与 StripedHyena2 和 Transformer++ 架构进行了比较。结果表明,dnaHNet 实现了卓越的计算和推理效率,在困惑度扩展以及蛋白质变体效应和基因必需性预测的下游性能方面始终优于基准模型。此外,该模型展示了一种在无监督情况下学习生物学层级结构的涌现能力,通过其分层压缩机制有效地发现了密码子结构和功能基因组区域。
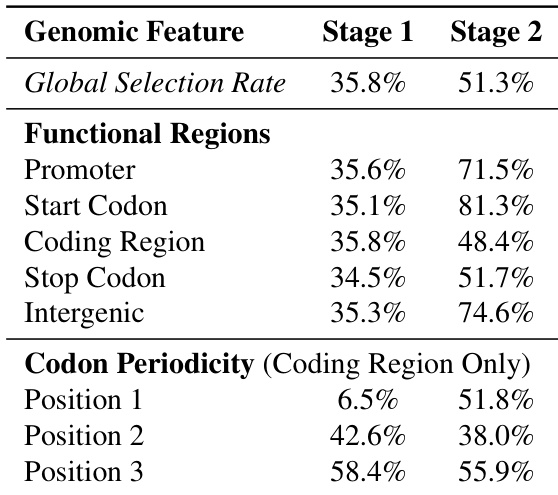
通过分析 dnaHNet 模型在不同阶段学习到的分层分块边界,研究发现第一阶段捕获了局部的三联体密码子结构,而第二阶段识别了更广泛的功能基因组组织。第一阶段在编码区表现出强烈的周期性,选择率随密码子位置显著变化。与编码区相比,第二阶段对启动子和基因间区等功能区域表现出更高的选择率。整体全局选择率从第一阶段到第二阶段有所增加。
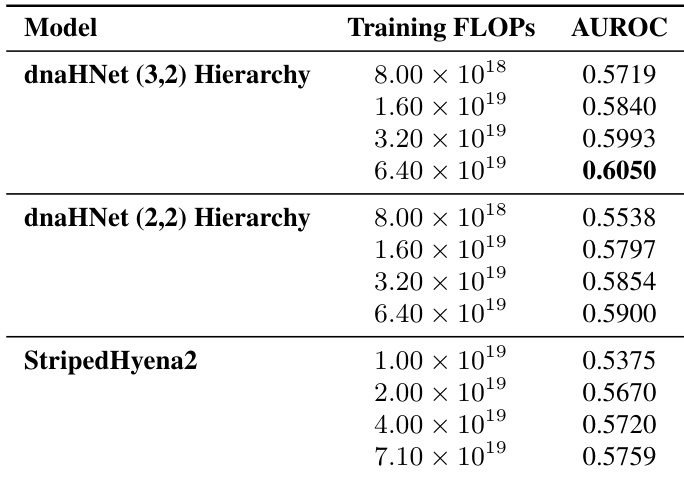
在各种训练计算预算下,评估了 dnaHNet 相对于 StripedHyena2 和 Transformer 基准的 zero-shot 蛋白质变体效应预测性能。结果显示,随着训练 FLOPs 的增加,dnaHNet 在预测实验适应性方面始终获得更高的 Spearman 相关性。与 StripedHyena2 和 Transformer 架构相比,dnaHNet 展示了更优的预测准确性。随着训练计算量的增加,dnaHNet 的预测性能稳步提高。在更高的训练计算规模下,dnaHNet 与基准模型之间的性能差距进一步扩大。
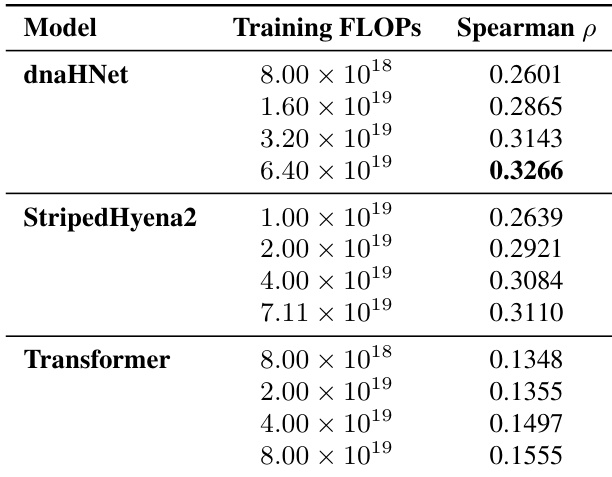
在各种计算预算下,比较了两种 dnaHNet 分层配置与 StripedHyena2 基准在基因必需性预测性能方面的表现。结果显示,随着训练计算量的增加,两种 dnaHNet 架构在 AUROC 方面始终优于基准。随着训练 FLOPs 的增加,两个 dnaHNet 层级版本都表现出预测准确性的提升。在匹配的计算水平下,dnaHNet (3,2) 层级比 (2,2) 层级实现了更高的 AUROC 分数。在测试的计算范围内,dnaHNet 模型相对于 StripedHyena2 保持着性能优势。
通过分析分层分块边界并测试其对蛋白质变体效应和基因必需性的预测能力,对 dnaHNet 模型进行了评估。研究结果表明,该模型通过其多阶段层级结构成功捕获了局部密码子结构和更广泛的功能基因组组织。在各种计算预算下,dnaHNet 始终优于 StripedHyena2 和 Transformer 基准,在蛋白质适应性和基因必需性任务中均展示了卓越的扩展性和预测准确性。