Command Palette
Search for a command to run...
SeedFold:扩展生物分子结构预测
SeedFold:扩展生物分子结构预测
Yi Zhou Chan Lu Yiming Ma Wei Qu Fei Ye Kexin Zhang Lan Wang Minrui Gui Quanquan Gu
摘要
高精度的生物分子结构预测是构建生物分子基础模型的关键组成部分,而构建基础模型中最关键的方面之一,便是确定模型扩展的“配方”。在本工作中,我们提出了SeedFold,一种能够有效提升模型容量的折叠模型。我们的贡献主要体现在三个方面:首先,我们提出了一种高效的Pairformer宽度扩展策略,以增强模型的表征能力;其次,我们引入了一种新型的线性三角注意力机制,显著降低计算复杂度,从而实现高效扩展;最后,我们构建了一个大规模的知识蒸馏数据集,大幅扩充了训练数据规模。在FoldBench上的实验结果表明,SeedFold在大多数蛋白质相关任务中表现优于AlphaFold3。
一句话总结
字节跳动种子团队提出 SeedFold,一种可扩展的生物分子结构预测模型,通过扩大 Pairformer 的宽度提升模型容量,采用线性三角注意力机制降低计算复杂度,并利用包含 2650 万样本的蒸馏数据集,在 FoldBench 上达到最先进性能,且在蛋白质相关任务上超越 AlphaFold3。
主要贡献
- 本文针对生物分子结构预测模型的可扩展性挑战,识别出 Pairformer 宽度扩展是提升表征能力的关键策略,突破了现有架构中固定隐藏维度带来的瓶颈。
- 提出一种新颖的线性三角注意力机制,将计算复杂度从立方级降低至二次级,实现高效扩展的同时不牺牲预测精度。
- 构建了一个大规模的 2650 万样本蒸馏数据集,源自 AlphaFold2,显著扩充了训练数据并提升模型泛化能力,使 SeedFold 在 FoldBench 基准测试中多数蛋白质相关任务上超越 AlphaFold3。
引言
作者利用大规模模型扩展的最新进展,提升生物分子结构预测这一药物发现与结构生物学关键任务的性能。尽管先前模型如 AlphaFold2 和 AlphaFold3 已实现高精度,但受限于固定架构设计——尤其是狭窄的成对表示和计算成本高昂的三角注意力机制——导致模型容量与可扩展性受限。SeedFold 的主要贡献在于三方面:首先,将 Pairformer 宽度扩展至 512,显著提升表征能力;其次,引入线性三角注意力机制,将计算复杂度从立方级降至二次级,实现高效扩展;第三,构建了一个包含 2650 万样本的大型蒸馏数据集,源自 AlphaFold2,以增强训练数据的质量与多样性。在 FoldBench 上的实验表明,SeedFold 在多个蛋白质相关任务中超越 AlphaFold3 及其他开源模型,不同注意力变体在不同相互作用类型上展现出各自优势。
数据集

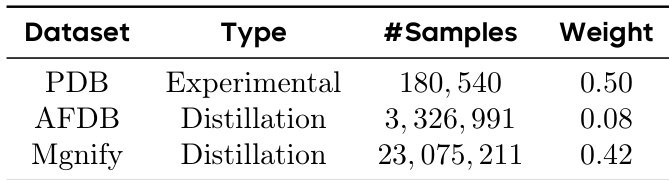
- SeedFold 的数据集包含 2650 万样本,通过从两个主要来源进行大规模数据蒸馏扩展:实验数据集(0.18M)和源自 AFDB 与 MGnify 的蒸馏数据集。
- 实验数据集包含截至 2021 年 9 月 30 日的 PDB 结构,经 Bolt 的处理流程处理,对链和界面进行聚类;训练过程中,样本按分子类型和聚类大小加权,以确保均衡表示。
- AFDB 数据集贡献了 330 万条来自 AlphaFold2 对 UniProt 序列预测的结构。序列在 50% 一致性水平上聚类,pLDDT 低于 0.8 的结构被过滤掉。短单体(少于 200 个残基)以 0.08 的概率采样,随后在长度范围内进行均匀采样。
- MGnify 数据集构成蒸馏数据的核心,包含来自环境微生物的 2300 万条宏基因组序列。少于 200 个残基的序列被移除,随后使用 MMSecs 在 30% 一致性水平上聚类。多序列比对(MSA)通过 colabfold_search 生成,使用 uniref30_db 和 colabfold_env 数据库。高质量结构通过 OpenFold 与 AlphaFold2 官方权重推断得出。
- AFDB 数据集偏向短蛋白质(中位长度 95),而 MGnify 包含更多样化且更长的序列(中位长度 435),对建模长蛋白质结构具有重要价值。
- 训练过程中,模型以相等概率从实验数据集和蒸馏数据集中采样。蒸馏数据用于持续训练阶段,移除后导致内部蛋白质 lDDT 显著下降,凸显其在模型性能中的关键作用。
- 未采用显式裁剪;而是通过基于长度的采样策略控制输入大小,预处理阶段构建了序列长度、聚类 ID 和 pLDDT 分数等元数据,以支持训练加权与过滤。
方法
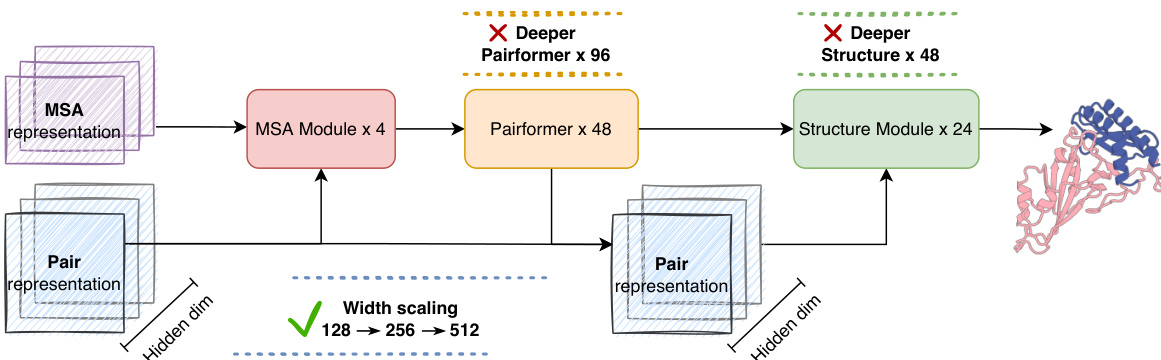
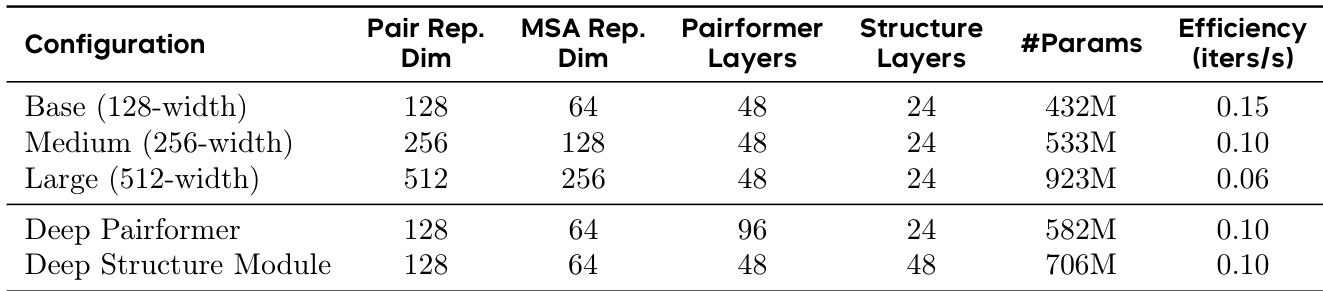
作者采用模块化架构构建其蛋白质折叠模型,围绕主干(trunk)和结构模块展开。主干模块负责编码进化与结构信息,由 MSA 模块和 Pairformer 模块组成。MSA 模块在多轮迭代中处理多序列比对(MSA),提取进化特征并更新成对表示。该成对表示随后用于更新 MSA 隐藏表示,形成双向信息流。Pairformer 模块进一步通过成对三角乘法和三角注意力精炼成对表示,捕捉令牌间相互作用。结构模块以主干输出的编码成对与单个表示为条件,执行全原子结构生成。整体框架旨在探索不同扩展策略,模型容量主要受限于成对表示的隐藏维度。作者研究了主干宽度、主干深度和结构模块深度的扩展,以确定提升性能的最优路径。
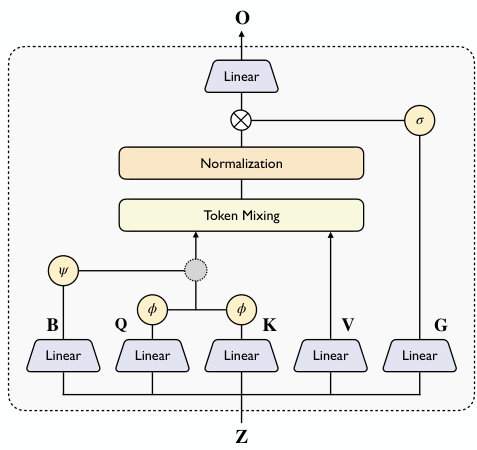
为解决三角注意力操作的计算瓶颈,作者提出一种新型线性三角注意力模块,作为原始三角注意力的即插即用替代方案。该模块旨在通过引入线性注意力(一种节省内存的技术)降低复杂度。标准三角注意力操作通过为每行和每列计算注意力分数来更新成对表示 Z∈Rn×n×d,需显式生成一个 n×n 的大矩阵,导致复杂度为 O(n3d)。线性注意力重构将 softmax 核替换为非线性特征映射,利用右乘技巧将计算成本从 O(n2d) 降低至 O(nd2)。然而,原始线性注意力未包含偏置项,而该偏置项对三维空间中的几何推理至关重要。作者提出一种三角形式的线性注意力,通过引入偏置项 B 重新加权注意力分数。他们提出两种变体:加性线性三角注意力(Additive Linear Triangular Attention),使用加法操作对注意力分数进行加权或降权;门控线性三角注意力(Gated Linear Triangular Attention),使用乘法操作作为门控机制控制信息流。该模块的架构如以下图示所示。
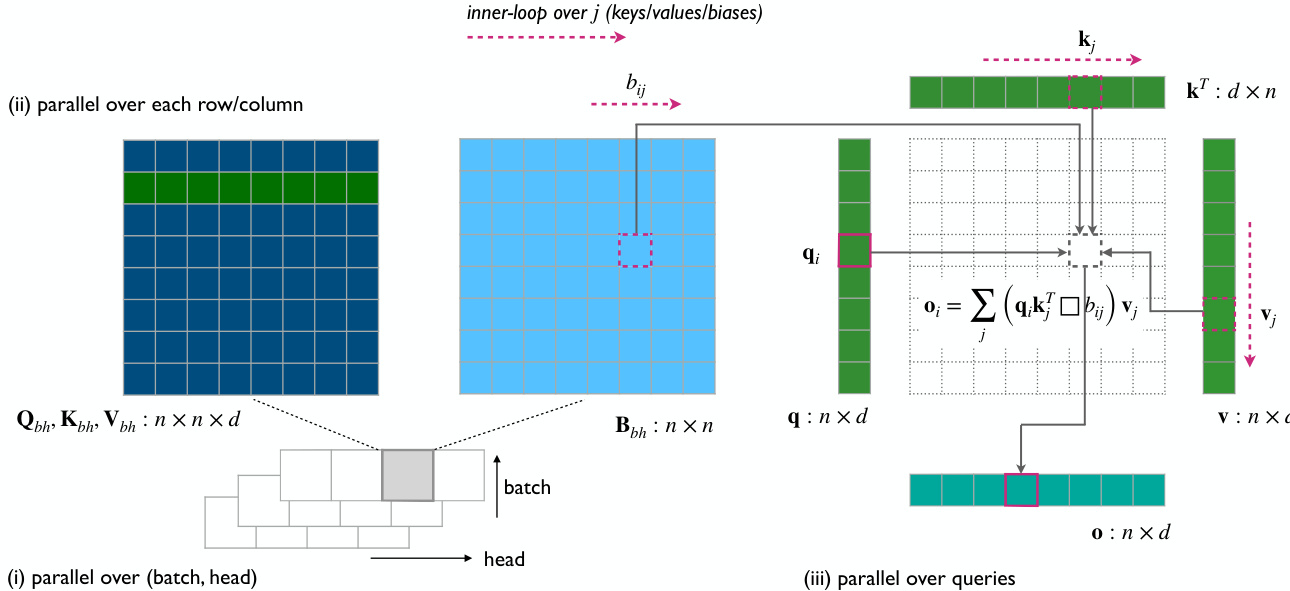
门控线性三角注意力变体采用逐点矩阵乘法,破坏了矩阵链乘积结构,导致无法使用右乘技巧。为缓解此问题,作者开发了一种针对 CUDA 设备优化的分块线性注意力版本,以降低内存占用。该模块的实现进一步在 Triton 内核中详细说明,用于计算输出 O 及其梯度。内核将批量和头维度合并为单一批量维度,并在大小为 n 的第一维度上并行化,迭代第二维度以聚合结果,避免显式生成完整的 n×n 矩阵。线性三角注意力模块的架构包含输出上的归一化与门控步骤,遵循 Lightning Attention 设计,该设计在大语言模型中已被证明有效。以下图示展示了该内核的实现。
实验
- 两阶段训练策略验证了模型性能的提升:第一阶段(384-token 裁剪,64 扩散批量)实现快速探索,第二阶段(640-token 裁剪,32 扩散批量)增强长序列建模能力,在 FoldBench 上取得最先进结果。
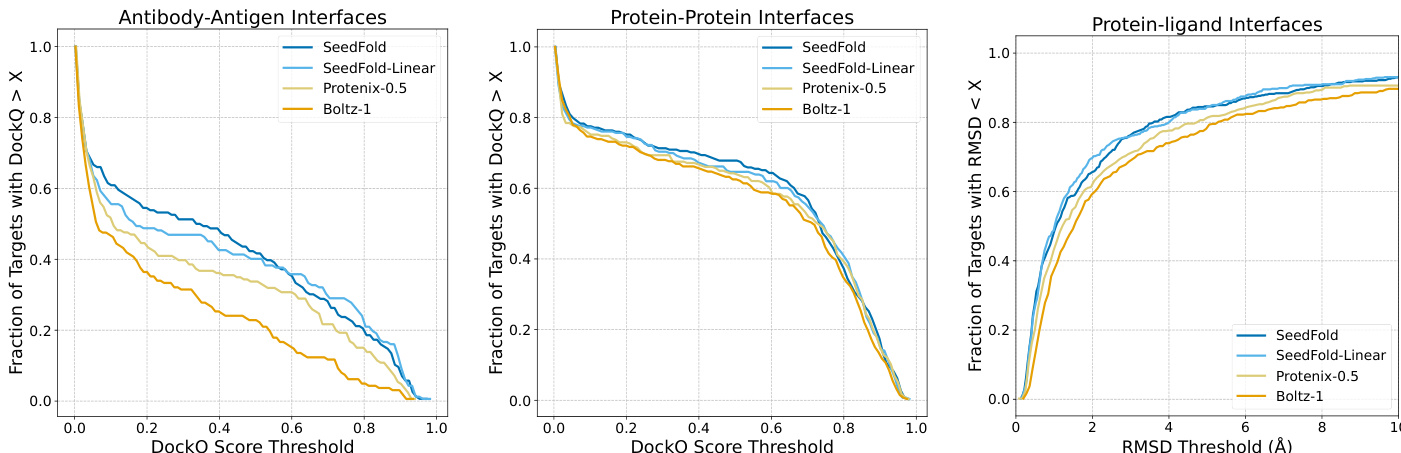
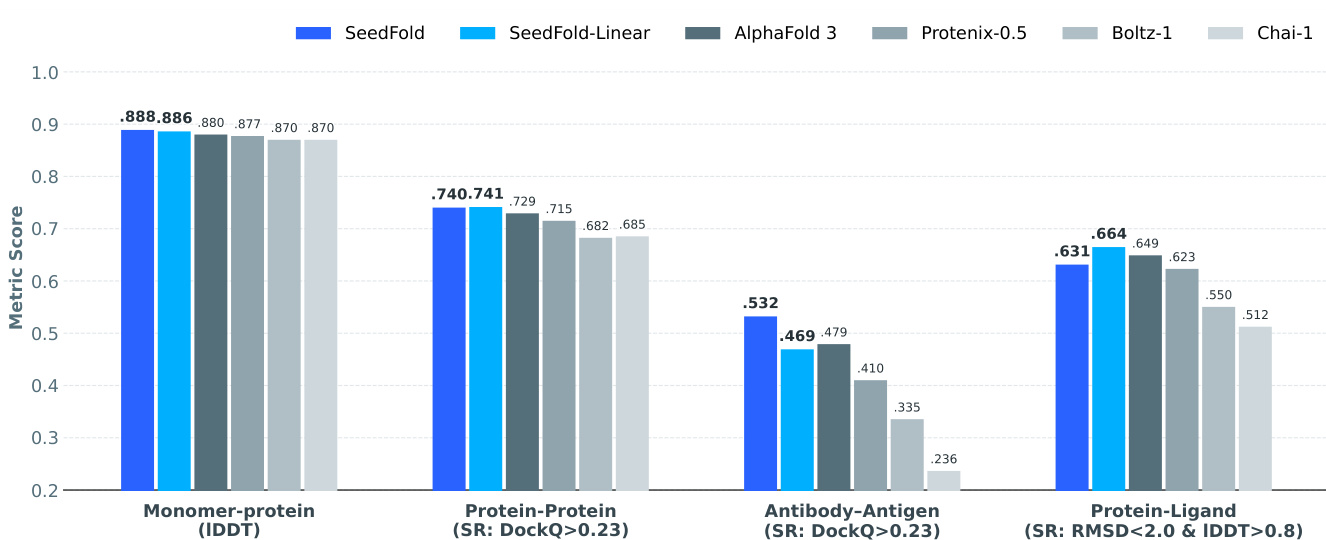
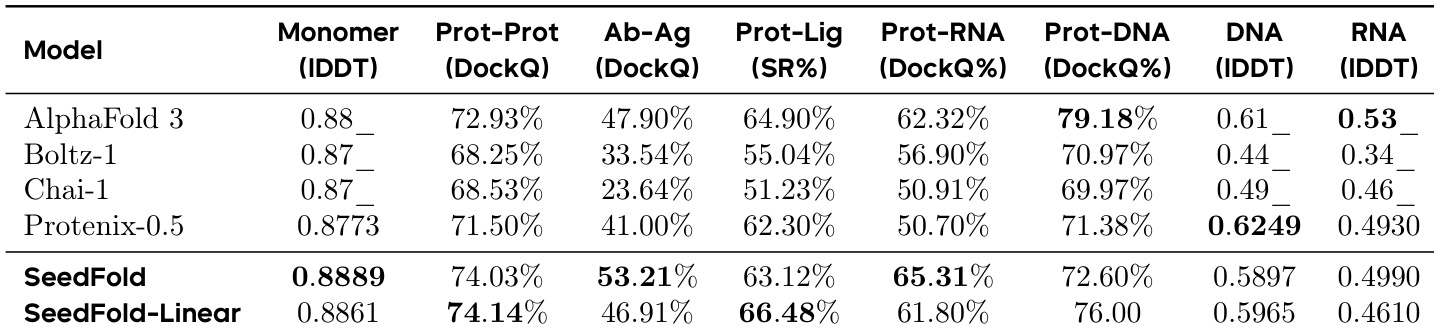
- SeedFold(512 宽度,原始三角注意力)在单体上达到 0.8889 lDDT,在抗体-抗原界面达到 53.21% DockQ,优于 AlphaFold3;SeedFold-Linear(384 宽度,GatedLinearTriAtt)在蛋白-配体(66.48% 成功)和蛋白-蛋白界面(74.14% DockQ)任务中领先。
- 消融实验确认门控线性三角注意力在核酸相关任务(蛋白-RNA/DNA)和界面预测中表现更优,持续优于加性线性三角注意力与原始注意力。
- 单体蒸馏数据至关重要:移除后导致内部蛋白质结构精度立即下降,凸显其在维持模型鲁棒性中的关键作用。
- 训练稳定性技术——延长预热(3000 步)与降低学习率(0.001)——使大模型成功收敛,优化后的 Large 模型展现出更稳定的训练动态与优于 Large-Raw 的验证性能。
作者采用两阶段训练策略与精度感知优化训练 SeedFold 模型,在 FoldBench 上实现最先进性能。结果表明,SeedFold 在蛋白质单体预测与抗体-抗原界面预测中树立新基准,而 SeedFold-Linear 在蛋白-配体与蛋白-蛋白界面预测中领先,充分验证了宽度扩展与线性注意力机制的有效性。
作者采用加权采样策略训练数据,AFDDB 与 Mgnify 蒸馏数据集贡献的样本远多于 PDB 实验数据集。结果表明,模型性能受各数据集相对权重影响,对蒸馏数据赋予更高权重,以强化从结构化、高质量样本中学习的能力。
结果表明,SeedFold 与 SeedFold-Linear 在大多数 FoldBench 任务中达到最先进或极具竞争力的性能,SeedFold 在蛋白质单体与抗体-抗原界面预测中领先,而 SeedFold-Linear 在蛋白-配体与蛋白-蛋白界面预测中表现卓越。两模型持续优于 AlphaFold3 及其他开源方法,充分证明宽度扩展与 GatedLinearTriAtt 使用在提升精度与效率方面的有效性。
作者采用扩展策略提升模型容量,结果显示,更大宽度与更多参数的模型性能更优,如从 Base 到 Large 配置的转变所示。结果表明,增加 Pairformer 宽度显著影响效率,Large 模型每秒迭代次数低于较小变体,而 Deep Pairformer 与 Deep Structure Module 配置虽在计算速度上有所牺牲,但显著提升了模型容量。
结果表明,SeedFold 在抗体-抗原界面预测中表现卓越,全面超越其他所有模型,覆盖整个 DockQ 评分范围。在蛋白-蛋白与蛋白-配体界面预测中,SeedFold 与 SeedFold-Linear 均持续优于 Protenix-0.5 与 Boltz-1,其中 SeedFold-Linear 在蛋白-配体任务中取得最佳表现。