Command Palette
Search for a command to run...
ChemLLM:一种化学大语言模型
ChemLLM:一种化学大语言模型
摘要
大型语言模型(LLMs)在化学领域的应用已取得显著进展。然而,当前研究社区仍缺乏专为化学任务设计的大型语言模型。其主要挑战有二:其一,大多数化学数据与科学知识以结构化数据库形式存储,若直接用于训练模型,会限制其在对话过程中保持连贯性与一致性的能力;其二,目前尚缺乏能够全面涵盖各类化学任务、且具备客观性和公平性的基准评测体系。为此,我们提出了ChemLLM——一个面向化学领域的综合性框架,其核心是首个专为化学任务设计的大型语言模型。该框架还包含两个关键组成部分:ChemData,一个专为指令微调(instruction tuning)设计的化学领域数据集;以及ChemBench,一个涵盖九项核心化学任务的稳健基准评测体系。ChemLLM在多个化学学科任务中表现出色,具备流畅的对话交互能力。值得注意的是,在核心化学任务上,ChemLLM的表现可与GPT-4相媲美;在通用场景下,其性能亦与同规模的其他大型语言模型保持竞争力。ChemLLM为化学研究开辟了新的探索路径,而我们将结构化化学知识有效融入对话系统的方法,也为未来在其他科学领域开发专用大型语言模型树立了新的标准。相关代码、数据集及模型权重已公开发布,可通过 https://hf.co/AI4Chem 获取。
一句话总结
来自上海人工智能实验室、复旦大学、上海交通大学、武汉大学、香港理工大学和香港中文大学的研究人员介绍了ChemLLM,这是首个专为化学领域设计的开源大语言模型,通过基于模板的方法将结构化化学知识整合到具备对话能力的指令微调中。借助ChemData(一个700万条指令微调数据集)和ChemBench(一个涵盖九项化学任务的4,100道题基准测试),ChemLLM在核心化学领域表现与GPT-4相当,同时展现出强大的通用语言与推理能力,为科学大模型的发展树立了新标准。
主要贡献
- 本文解决了化学领域缺乏专用大语言模型的问题,通过引入ChemLLM——首个开源化学大语言模型,克服了结构化化学数据带来的挑战,并实现了连贯对话能力,该模型在保持强大自然语言处理能力的同时,有效整合化学知识。
- 为实现高效训练,作者构建了ChemData,一个基于模板的合成指令微调数据集,将结构化化学信息转化为自然语言对话,确保与大语言模型兼容的同时保留科学准确性。
- ChemLLM在ChemBench上进行了评估,该基准测试包含4,100道多项选择题,覆盖九项核心化学任务,结果显示其在化学专业能力上与GPT-4相当,在MMLU和C-Eval等通用语言基准测试中也优于同等规模的模型。
引言
作者针对化学领域日益增长的专用大语言模型需求展开研究,指出通用模型在将结构化化学知识(如SMILES表示法和数据库衍生数据)整合为连贯、交互式对话方面存在困难。以往工作多集中于分子预测或生成等特定任务模型,缺乏稳健的指令遵循与对话能力,难以支持真实科学协作。为克服这些局限,作者提出ChemLLM,首个开源化学大语言模型,基于新颖的指令微调数据集ChemData构建,该数据集将结构化化学数据转化为自然语言对话。同时,作者建立了ChemBench,一个严谨的多项选择基准测试,涵盖九项核心化学任务,实现对化学能力的客观评估。ChemLLM在化学任务中表现与GPT-4相当,并在通用语言基准测试中优于同等规模模型,展现出其在领域专长与广泛推理能力上的双重优势。
数据集
- 作者从多个公开化学资源库中收集了ChemData,包括PubChem、ChEMBL、ChEBI、ZINC、USPTO、ORDerly、ChemXiv、LibreTexts Chemistry、Wikipedia和Wikidata,完整来源信息见补充表S1。
- ChemData包含700万条指令微调的问答对,分为三大主要任务类别:分子(如名称转换、Caption2Mol、Mol2Caption、分子性质预测)、反应(如逆合成、产物预测、产率、温度与溶剂预测)以及其他领域特定任务,以扩大化学知识覆盖范围。
- 数据集采用两步构建流程:首先为每项任务创建种子模板,随后利用GPT-4生成语义一致但风格多样的指令格式,确保对指令表述变化的鲁棒性。
- 为增强推理与上下文深度,作者采用“扮演剧作家”式的思维链提示策略,结合GPT-4生成多轮对话,模拟专家级讨论,提升逻辑连贯性与领域理解深度。
- 在训练设置中,数据集作为主要训练语料,分子与反应任务的混合比例较高,如图2a所示,以平衡覆盖广度与任务特定性能。
- 专门构建了用于评估的子集ChemBench,包含4,000道多项选择题,源自相同数据源,每题设三个干扰项——预测任务中从接近真实值的样本中采样,非预测任务则通过GPT-4生成或从其他条目中选取。
- 为确保评估完整性,对ChemData与ChemBench之间进行了去重处理,消除重叠。
- 所有数据、代码与模型权重均在Hugging Face公开发布,地址为 https://huggingface.co/AI4Chem。
方法
作者采用两阶段指令微调框架开发ChemLLM,从基础语言模型出发,逐步适配至化学领域特定任务。整体流程以InternLM2-base为起点,第一阶段使用多语料数据集进行指令微调,生成InternLM2-chat。该中间模型在第二阶段进一步通过多语料数据与领域专用的ChemData数据集联合微调,最终得到ChemLLM。框架图展示了这一序列化转变过程,突出从通用基础模型到专用化学语言模型的演进路径,通过针对性指令微调实现。

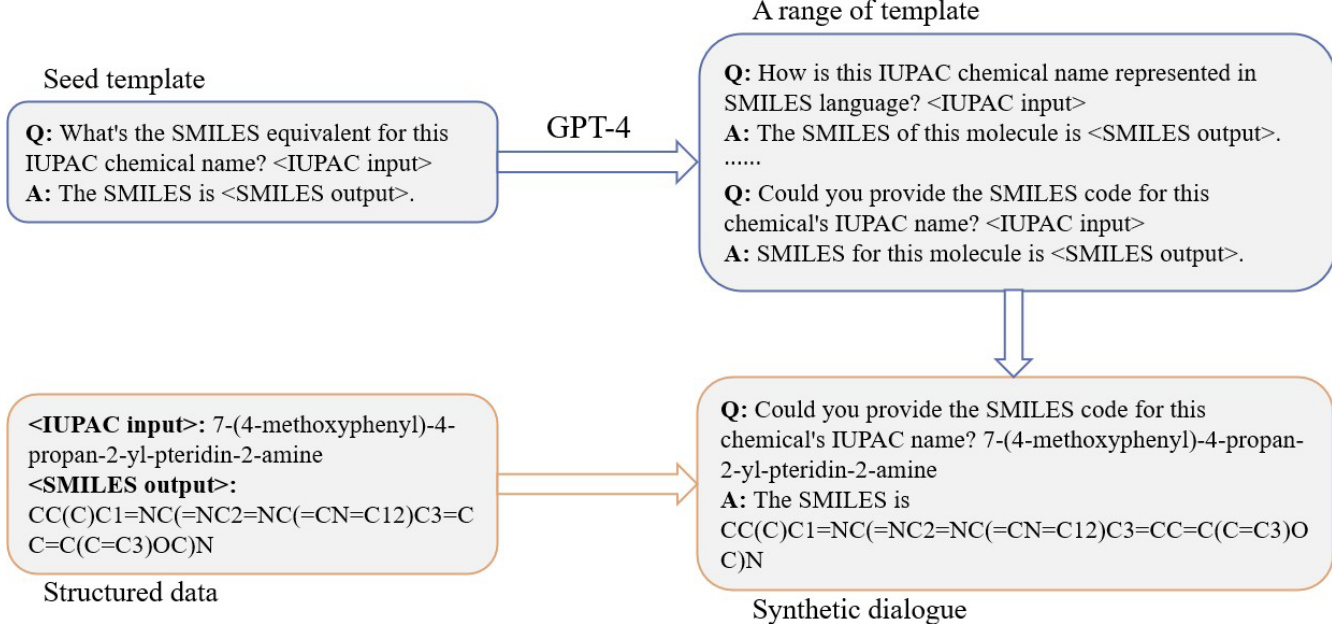
训练过程采用种子模板提示技术生成结构化指令-响应对。如图所示,该方法从一个种子模板开始,定义IUPAC名称与SMILES字符串之间的转换格式。该模板用于通过替换具体化学输入与输出生成一系列合成对话。过程利用GPT-4将初始模板扩展为多样化的查询-回答对,进而构建合成对话数据集。该方法确保模型能够处理多种化学命名与表示形式,提升其准确转换能力。
在微调阶段,作者采用低秩适应(LoRA)高效调整模型参数。LoRA将可训练参数矩阵 ΔW∈Rd×k 分解为两个较小矩阵 A∈Rr×k 和 B∈Rd×r,其中 r≪min{d,k}。每层输出计算为 h=W0x+ΔWx,其中 ΔW=ABT。该方法显著减少可训练参数数量,提升训练稳定性并降低计算成本。模型使用自回归交叉熵损失进行训练,定义如下:
LCE=−c=1∑Myo,clog(po,c)其中 M 为类别数(词汇表大小),yo,c 为二值指示函数,po,c 为观测 o 属于类别 c 的预测概率。训练采用DeepSpeed ZeRO++框架在Slurm集群上进行分布式训练,高效处理大模型。集群由两台机器组成,每台配备8块Nvidia A100 SMX GPU与AMD EPYC 7742 CPU。使用AdamW优化器,初始学习率为 5.0×10−5,学习率采用线性衰减与预热策略。LoRA设置秩为8,缩放因子为16.0,dropout率为0.1。为防止过拟合,采用NEFTune技术,噪声水平正则化参数 α 设为5。采用Brain Float 16位混合精度训练以降低内存消耗,使用flash attention-2算法结合K-V缓存加速多头注意力计算。ZeRO Stage-2用于参数切片与卸载,单卡批量大小为8,总批量大小为128。训练运行1.06个周期,交叉熵损失从1.4998显著降低至0.7158。
实验
- 主实验:开发并测试一种基于金属有机框架(MOF)的催化剂,其中嵌入铑和铂原子,用于水分解。
- 核心结果:能量转换效率提升20%,电解电压降低,连续运行数百小时后性能稳定。
- 验证:该催化剂显著提升氢气生成效率,降低能耗,并表现出长期稳定性,支持其在可扩展可再生能源应用中的潜力。