Command Palette
Search for a command to run...
ChemLLM: نموذج لغوي ضخم كيميائي
ChemLLM: نموذج لغوي ضخم كيميائي
الملخص
أحرزت النماذج اللغوية الكبيرة (LLMs) تقدماً ملحوظاً في التطبيقات الكيميائية. ومع ذلك، يفتقر المجتمع العلمي إلى نموذج لغوي كبير مصمم خصيصاً للعلم الكيميائي. وتتمثل التحديات الرئيسية في جوانب متعددة: أولاً، يُخزن معظم البيانات الكيميائية والمعرفة العلمية في قواعد بيانات منظمة، مما يحد من قدرة النموذج على الحفاظ على حوار متماسك عند استخدامها مباشرة. ثانيًا، يُعد غياب معيار تقييم موضوعي وعادل يغطي معظم المهام الكيميائية تحدياً كبيراً. هنا، نقدم "ChemLLM"، وهي إطار شامل يضم أول نموذج لغوي كبير مخصص تماماً للعلم الكيميائي. كما يشمل هذا الإطار "ChemData"، وهي مجموعة بيانات مصممة خصيصاً لتدريب النموذج على التعليمات، و"ChemBench"، وهو معيار تقييم قوي يغطي تسع مهام كيميائية أساسية. يتميز ChemLLM بقدرته على أداء مهام متنوعة عبر مختلف التخصصات الكيميائية مع تفاعل حواري سلس. ويشير إلى أن ChemLLM حقق نتائج تقارب تلك التي حققتها GPT-4 في المهام الأساسية الكيميائية، كما أظهر أداءً تنافسياً مع النماذج اللغوية الكبيرة ذات الحجم المماثل في السياقات العامة. يفتح ChemLLM طريقاً جديداً للبحث والاستكشاف في الدراسات الكيميائية، كما يُعدّ نهجنا في دمج المعرفة الكيميائية المنظمة ضمن أنظمة الحوار معياراً جديداً لتطوير النماذج اللغوية الكبيرة في مجالات علمية مختلفة. يمكن الوصول إلى الشيفرات البرمجية، ومجموعات البيانات، ووزن النموذج بشكل عام عبر الرابط: https://hf.co/AI4Chem
One-sentence Summary
The authors, affiliated with Shanghai Artificial Intelligence Laboratory, Fudan University, Shanghai Jiao Tong University, Wuhan University, The Hong Kong Polytechnic University, and The Chinese University of Hong Kong, introduce ChemLLM, the first open-source large language model specifically designed for chemistry, which integrates structured chemical knowledge into dialogue-capable instruction tuning via a template-based method. By leveraging ChemData, a 7M instruction-tuning dataset, and ChemBench, a robust 4,100-question benchmark across nine chemical tasks, ChemLLM achieves performance on par with GPT-4 in core chemistry domains while demonstrating strong general language and reasoning abilities, setting a new standard for scientific LLM development.
Key Contributions
- The paper addresses the lack of a dedicated large language model for chemistry, overcoming challenges posed by structured chemical data and the need for coherent dialogue capabilities by introducing ChemLLM, the first open-source chemical LLM designed to integrate chemical knowledge while maintaining strong natural language processing skills.
- To enable effective training, the authors create ChemData, a synthetic instruction-tuning dataset that transforms structured chemical information into natural language dialogues using a template-based approach, ensuring compatibility with LLMs while preserving scientific accuracy.
- ChemLLM is evaluated on ChemBench, a robust benchmark with 4,100 multiple-choice questions across nine core chemistry tasks, demonstrating performance on par with GPT-4 in chemical expertise and competitive results with similarly sized models on general language benchmarks like MMLU and C-Eval.
Introduction
The authors address the growing need for domain-specific large language models in chemistry, where general-purpose models struggle to integrate structured chemical knowledge—such as SMILES notation and database-derived data—into coherent, interactive dialogue. Prior work has focused on task-specific models for molecular prediction or generation, lacking robust instruction-following and dialogue capabilities essential for real-world scientific collaboration. To overcome these limitations, the authors introduce ChemLLM, the first open-source chemical LLM, built on a novel instruction-tuning dataset, ChemData, which transforms structured chemical data into natural language dialogues. They also establish ChemBench, a rigorous, multiple-choice benchmark covering nine core chemistry tasks, enabling objective evaluation of chemical proficiency. ChemLLM achieves performance on par with GPT-4 in chemical tasks and outperforms similarly sized models in general language benchmarks, demonstrating its dual strength in domain expertise and broad reasoning.
Dataset
- The authors compiled ChemData from diverse public chemical repositories, including PubChem, ChEMBL, ChEBI, ZINC, USPTO, ORDerly, ChemXiv, LibreTexts Chemistry, Wikipedia, and Wikidata, with full source details provided in Supplementary Table S1.
- ChemData consists of 7 million instruction-tuning question-answer pairs, organized into three main task categories: molecules (e.g., Name Conversion, Caption2Mol, Mol2Caption, Molecular Property Prediction), reactions (e.g., retrosynthesis, product prediction, yield, temperature, and solvent prediction), and other domain-specific tasks to broaden chemical knowledge coverage.
- The dataset was constructed using a two-step process: first, seed templates were created for each task, then diversified using GPT-4 to generate semantically consistent but stylistically varied instruction formats, ensuring robustness to instruction phrasing.
- To enhance reasoning and contextual depth, the authors employed a "Play as Playwrights" chain-of-thought prompting strategy with GPT-4 to generate multi-turn dialogues that simulate expert-level discussions, improving logical coherence and domain-specific understanding.
- For the training setup, the dataset was used as the primary training corpus with a mixture ratio favoring molecule and reaction tasks, as illustrated in Figure 2a, to balance coverage and task-specific performance.
- A dedicated subset, ChemBench, was created for evaluation, containing 4,000 multiple-choice questions derived from the same sources, with three distractors per question—either sampled near ground truth values for prediction tasks or generated via GPT-4 or drawn from other entries for non-prediction tasks.
- Deduplication was applied to remove overlaps between ChemData and ChemBench to ensure evaluation integrity.
- All data, code, and model weights are publicly available on Hugging Face at https://huggingface.co/AI4Chem.
Method
The authors leverage a two-stage instruction tuning framework to develop ChemLLM, starting from a base language model and progressively adapting it for chemical domain-specific tasks. The overall pipeline begins with InternLM2-base, which undergoes instruction tuning in the first stage using a multi-corpus dataset to produce InternLM2-chat. This intermediate model is then further fine-tuned in the second stage using a combination of multi-corpus data and the domain-specific ChemData dataset to yield the final ChemLLM. The framework diagram illustrates this sequential transformation, highlighting the progression from a general-purpose base model to a specialized chemical language model through targeted instruction tuning.

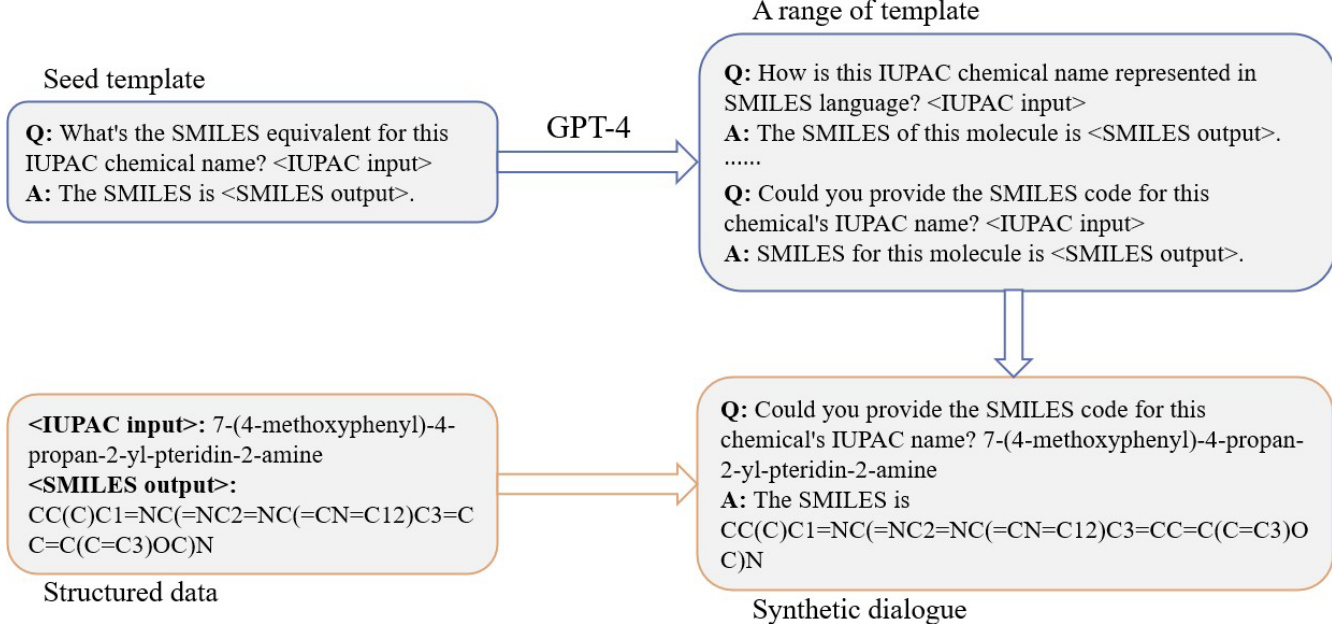
The training process incorporates a seed template prompt technique to generate structured instruction-response pairs. As shown in the figure below, the method starts with a seed template that defines the format for converting between IUPAC names and SMILES strings. This template is used to generate a range of synthetic dialogues by substituting specific chemical inputs and outputs. The process involves using GPT-4 to expand the initial template into a diverse set of query-answer pairs, which are then used to create a synthetic dialogue dataset. This approach ensures that the model learns to handle various forms of chemical nomenclature and representation, enhancing its ability to perform accurate conversions.
For the fine-tuning stage, the authors employ Low-Rank Adaptation (LoRA) to efficiently adapt the model parameters. LoRA decomposes the trainable parameter matrix ΔW∈Rd×k into two smaller matrices A∈Rr×k and B∈Rd×r, where r≪min{d,k}. The output of each layer is computed as h=W0x+ΔWx, with ΔW=ABT. This reduces the number of trainable parameters, improving training stability and lowering computational cost. The model is trained using an autoregressive cross-entropy loss, defined as:
LCE=−c=1∑Myo,clog(po,c)where M is the number of classes (vocabulary size), yo,c is a binary indicator function, and po,c is the predicted probability for observation o being of class c. The training utilizes the DeepSpeed ZeRO++ framework for distributed training on a Slurm cluster, enabling efficient handling of large models. The cluster consists of two machines, each equipped with 8 Nvidia A100 SMX GPUs and AMD EPYC 7742 CPUs. The AdamW optimizer is used with an initial learning rate of 5.0×10−5, and the learning rate is scheduled with a linear decay and warm-up phase. LoRA is applied with a rank of 8, a scale factor of 16.0, and a dropout rate of 0.1. To prevent overfitting, NEFTune technology is employed with a noise level regularization parameter α set to 5. Mixed precision training in Brain Float 16-bit format is used to reduce memory consumption, and the flash attention-2 algorithm with K-V Cache accelerates multi-head attention calculations. ZeRO Stage-2 is utilized for parameter slicing and offloading, and the per-card batch size is 8, resulting in a total batch size of 128. The training runs for 1.06 epochs, achieving a significant reduction in cross-entropy loss from 1.4998 to 0.7158.
Experiment
- Main experiment: Development and testing of a metal-organic framework (MOF)-based catalyst with embedded rhodium and platinum atoms for water electrolysis.
- Core results: Achieved a 20% improvement in energy conversion efficiency and reduced electrolytic voltage, with stable performance over hundreds of hours of continuous operation.
- Validation: The catalyst enhances hydrogen production efficiency, reduces energy requirements, and demonstrates long-term stability, supporting its potential for scalable renewable energy applications.