Command Palette
Search for a command to run...
MolHIT:階層的離散拡散モデルによる分子グラフ生成の進展
MolHIT:階層的離散拡散モデルによる分子グラフ生成の進展
Hojung Jung Rodrigo Hormazabal Jaehyeong Jo Youngrok Park Kyunggeun Roh Se-Young Yun Sehui Han Dae-Woong Jeong
概要
拡散モデルを用いた分子生成は、AI駆動型ドラッグディスカバリーや材料科学において有望な研究方向性として浮上している。2次元分子グラフの離散的性質に適していることから、グラフ拡散モデルは広く採用されてきたが、既存のモデルは化学的妥当性が低く、1次元(1D)モデルと比較して望ましい物性を満たすことが困難であるという課題を抱えていた。本研究では、従来の手法に長年存在していた性能限界を克服する強力な分子グラフ生成フレームワーク「MolHIT」を提案する。MolHITは、化学的事前知識を追加カテゴリとして符号化する階層的離散拡散モデル(Hierarchical Discrete Diffusion Model)に基づいており、原子の化学的役割に応じて原子種を分離する「分離型原子符号化(decoupled atom encoding)」を導入している。全体として、MolHITはMOSESデータセットにおいて、グラフ拡散モデルとしては初めてほぼ完全な化学的妥当性を達成し、複数の指標において強力な1Dベースラインを上回る新しいSOTA(最先端)性能を達成した。さらに、マルチプロパティ制御生成やスケルトン拡張といった下流タスクにおいても優れた性能を示した。
One-sentence Summary
Researchers from KAIST AI, LG AI Research, and Seoul National University propose MolHIT, a hierarchical discrete diffusion model that enhances molecular graph generation by encoding chemical priors and decoupling atom roles, achieving near-perfect validity and SOTA performance on MOSES, with strong downstream applications in property-guided design and scaffold extension.
Key Contributions
- MolHIT introduces a Hierarchical Discrete Diffusion Model that encodes chemical priors through coarse-to-fine state transitions, addressing the low validity of prior graph diffusion models while preserving structural novelty.
- It proposes Decoupled Atom Encoding to split atom types by chemical roles like aromaticity and charge, resolving information loss in naive encodings and improving reconstruction and generation reliability.
- Evaluated on MOSES and other benchmarks, MolHIT achieves near-perfect validity and state-of-the-art performance across unconditional and conditional tasks, outperforming both 1D and existing 2D baselines.
Introduction
The authors leverage hierarchical discrete diffusion and chemically aware atom encoding to tackle molecular graph generation, where prior models struggle to balance validity and novelty. Existing graph diffusion approaches treat atoms as independent categories and use naive encodings that ignore chemical roles like aromaticity or charge, leading to invalid or unrealistic structures. MolHIT introduces a two-stage diffusion process that first learns coarse chemical identities before refining them, paired with Decoupled Atom Encoding that explicitly separates atom types by their functional roles—resulting in near-perfect validity on MOSES and outperforming both 1D and 2D baselines across benchmarks and downstream tasks like scaffold extension and multi-property generation.
Dataset
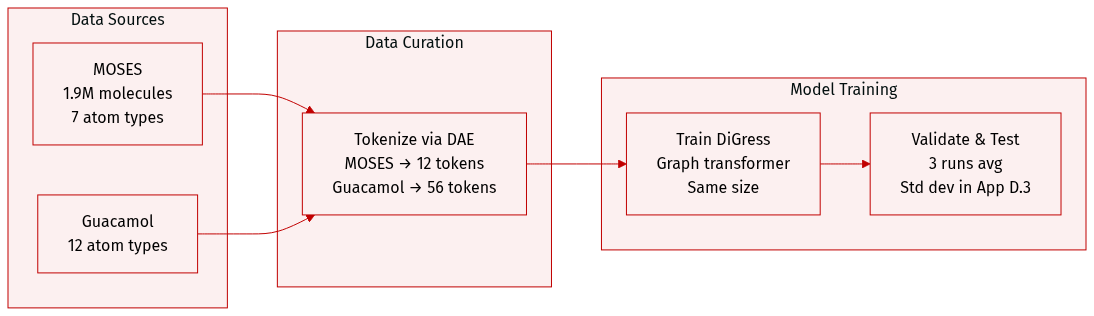
- The authors use two large molecular datasets: MOSES (1.9M molecules, 7 heavy atom types) and Guacamol (12 heavy atom types).
- Both datasets are processed via DAE: MOSES is augmented into 12 tokens, Guacamol is decoupled into 56 tokens.
- The model architecture follows DiGress, using the same graph transformer size.
- All results are averaged over three independent runs, with standard deviations in Appendix D.3.
Method
The authors leverage a Hierarchical Discrete Diffusion Model (HDDM) to generalize the standard discrete diffusion framework into a multi-stage corruption process, enabling more structured and chemically meaningful denoising for molecular graph generation. The core innovation lies in augmenting the state space with mid-level semantic categories that bridge the clean atom types and a final masked state, allowing the model to progressively refine predictions from broad chemical classes to specific atomic identities.
As shown in the framework diagram, the HDDM operates over a three-tiered state space: the clean state set S0, a set of mid-level states S1, and a single masked state S2={m}. The forward diffusion process is governed by a sequence of transition matrices that progressively map clean atoms into their semantic groups (e.g., halogens, aromatics, chalcogens, aliphatics) before ultimately transitioning to the masked state. This hierarchical structure is encoded via a block-structured transition kernel Q(1) that maps clean states to mid-level states, and Q(2) that maps all non-masked states to the mask. The cumulative forward transition at timestep t is defined as:
Qt=αtI+(βt−αt)Q(1)+(1−βt)Q(2),where αt and βt are monotonically decreasing diffusion schedules satisfying α0=β0=1 and αT=βT=0, with αt≤βt. This formulation ensures Chapman–Kolmogorov consistency, enabling tractable multi-step transitions.
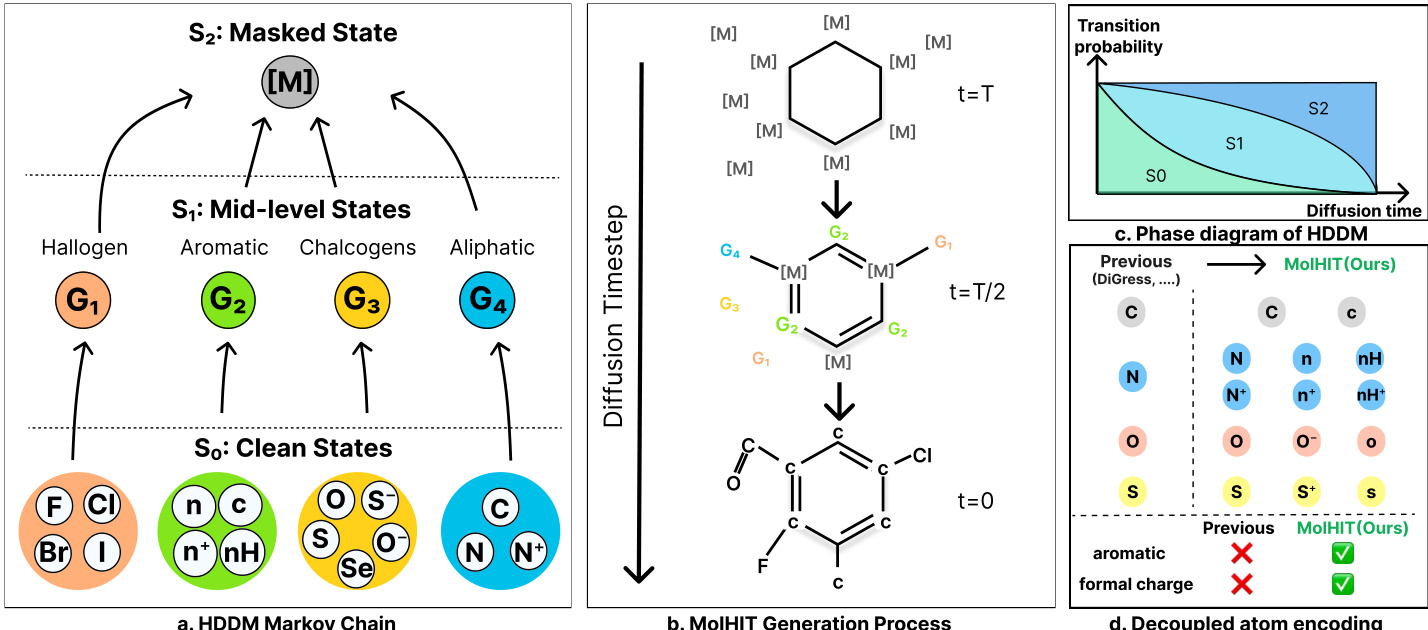
For molecular graph generation, the authors decouple the diffusion process for atoms and bonds. Atoms are perturbed via the HDDM process, while bonds follow a uniform transition kernel to ensure structural diversity. The forward dynamics are thus:
QX,t=αX,tI+(βX,t−αX,t)QX,t(1)+(1−βX,t)QX,t(2),QE,t=αE,tI+(1−αE,t)1dE1dET.The model is trained to predict the clean graph G0=(X0,E0) from a noisy graph Gt, using a cross-entropy loss that independently optimizes atom and bond predictions:
Lθ=Et,Gt∼q(⋅∣G0)[i=1∑n−logpθX(X0,i∣Gt,t)+λ1≤i<j≤n∑−logpθE(E0,ij∣Gt,t)],where λ balances node and edge contributions.
To enhance chemical fidelity, the authors introduce Decoupled Atom Encoding (DAE), which expands the atom vocabulary by explicitly encoding aromaticity, hydrogen saturation, and formal charge as distinct token states. This resolves structural ambiguities present in coarse encodings (e.g., distinguishing [n] from [nH]) and enables near-perfect reconstruction of complex motifs like heteroaromatics and zwitterions. As illustrated in the figure, DAE allows MolHIT to generate molecules with formal charges at proportions matching the training distribution, a capability absent in prior models.
Sampling is performed via a Project-and-Noise (PN) sampler, which projects the model’s denoised prediction onto the discrete manifold via categorical sampling and then re-noises it to the previous timestep using the forward kernel. This bypasses posterior constraints and encourages structural diversity. Temperature and top-p sampling are applied selectively to atom predictions to control the quality-diversity trade-off. The overall generation process, depicted in the figure, shows a molecule evolving from a fully masked state at t=T, through mid-level semantic states at t=T/2, to a fully reconstructed structure at t=0, guided by the hierarchical transition probabilities.
Experiment
- MolHIT achieves state-of-the-art performance on MOSES across key metrics including Quality, Validity, FCD, and Scaffold Novelty, demonstrating strong navigation of the drug-like chemical manifold while exploring novel structures.
- On GuacaMol, MolHIT outperforms baselines across most metrics despite using the full unfiltered dataset, showing robustness to charged and complex molecules; performance gaps in FCD are attributed to modeling challenges with extended atom vocabularies.
- In multi-property guided generation, MolHIT significantly improves conditioning precision (52.4% lower MAE) and reliability (Pearson r up to 0.950) without sacrificing validity, confirming effective control over chemical properties like QED, SA, MW, and logP.
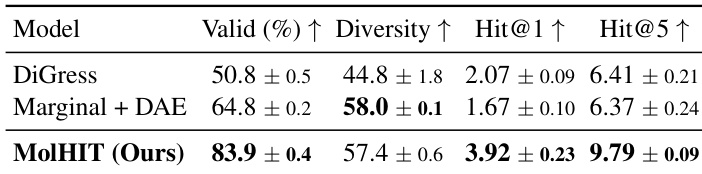
- For scaffold extension, MolHIT surpasses DiGress in validity, diversity, and hit rates, indicating superior ability to generate chemically plausible and structurally diverse extensions while preserving fixed scaffolds.
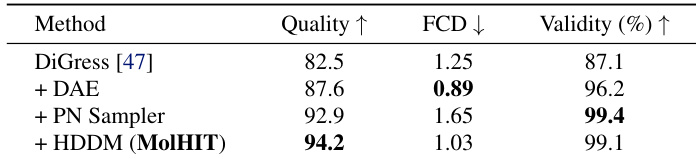
- Ablation studies confirm that DAE, PN Sampler, and HDDM each contribute meaningfully to overall performance, while temperature sampling reveals a trade-off between quality and novelty, with optimal settings yielding near-perfect validity and high quality.
The authors use MolHIT to generate molecules on the MOSES benchmark and compare it against both 1D sequence and 2D graph baselines. Results show MolHIT achieves the highest Quality and Scaffold Novelty while maintaining near-perfect validity, outperforming prior models in balancing structural innovation with chemical feasibility. The model also demonstrates strong distributional fidelity, as reflected in high Scaffold Retrieval and SNN scores, indicating it effectively captures the underlying drug-like chemical space without overfitting.
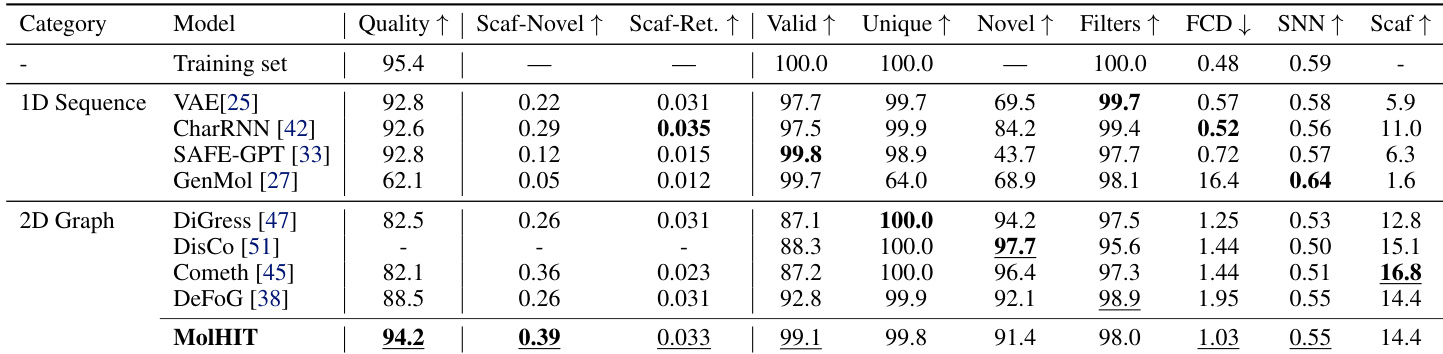
The authors use a full unfiltered GuacaMol dataset to evaluate MolHIT, contrasting with prior models trained on filtered subsets. Results show MolHIT achieves the highest validity and scaffold novelty while maintaining strong distributional fidelity, outperforming DiGress variants across most metrics despite using fewer training epochs. The model demonstrates robustness in handling charged atoms and broader chemical space without sacrificing structural quality.

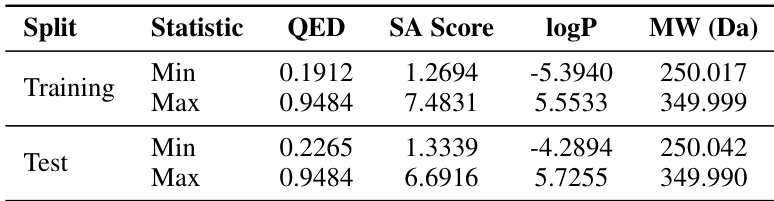
The authors use the MOSES dataset to evaluate multi-property guided generation, conditioning models on QED, SA, logP, and MW. Results show MolHIT achieves high precision in matching target properties with low MAE and strong Pearson correlation, while maintaining validity above 95%. This indicates the model effectively balances property control with structural feasibility.
The authors use an ablation study to show that integrating decoupled atom encoding, the PN sampler, and HDDM into DiGress progressively improves molecular generation quality, validity, and distributional fidelity. Results show that MolHIT achieves the highest Quality and near-perfect Validity while maintaining competitive FCD, indicating effective navigation of the drug-like chemical space. Each component contributes meaningfully, with the full MolHIT configuration outperforming all intermediate variants.
The authors evaluate MolHIT on scaffold extension tasks using the MOSES dataset, comparing it against DiGress and a marginal transition baseline with decoupled atom encoding. Results show MolHIT achieves significantly higher validity and Hit@1 and Hit@5 scores, indicating stronger capability to recover ground-truth molecular extensions while maintaining structural diversity. The improvements suggest MolHIT better balances fidelity to fixed scaffolds with exploration of valid chemical space.